One of the most severe ailments of the digestive system, which is genetic in nature, is diffuse polyposis, affecting the rectum and colon. This disease should be distinguished from damage to single and group polyps, since, affecting a person in his youth, by the age of 50, in almost 100 percent of cases it turns into oncology. The disease can occur in three main forms:
- Like diffuse familial polyposis;
- Gardner's syndrome;
- Peutz-Jeghers syndrome.
It is about the latter type that we will talk in more detail, in particular, we will find out what causes its development, consider the symptoms of the disease and methods of its treatment.
It's all due to heredity
Speaking about the reasons for the development of this syndrome, experts identify exclusively hereditary predisposition. The statistics are disappointing: if a person’s family has relatives who suffer from the disease, he will get sick with almost one hundred percent probability. More precisely, the disease will already be inherent in him from birth, and over time, as a rule, at a fairly young age, it will manifest itself.
It would seem that everything is quite clear. In fact, doctors consider this disease to be one of the most poorly understood. The thing is that so far researchers have been able to identify only one gene, the presence of which puts a person at risk. But doctors suggest the presence of a number of other genes, which have not yet been identified.
Peutz-Jeghers syndrome (Hamartoma polyposis)
Diagnosis is often difficult, due to the low prevalence of Peutz-Jeghers syndrome and the nonspecificity of dyspeptic symptoms. The disease can be suspected if the patient has pronounced pigmentation, which appears at an early age, or if there is evidence of a hereditary burden. A diagnostic search involves conducting a comprehensive examination of the digestive system. The most informative are:
- Abdominal sonography
. Non-invasive ultrasound scanning is used for rapid diagnosis of the syndrome. Ultrasound of the abdominal organs makes it possible to detect space-occupying formations in the intestinal lumen and pathology of other gastrointestinal organs. Sonography may not be informative enough for small polyps (up to 5 mm). - Contrast radiography.
Taking radiographs after oral administration of a thick barium suspension is necessary to identify polypoid growths. With Peutz-Jeghers anomaly, round-shaped filling defects are visible. Detection of unclear contours or local disturbance of peristalsis is unfavorable prognostically. - Endoscopic examination
. To visualize the mucous membrane of the upper parts of the digestive tube, an endoscopy is performed, and a colonoscopy is performed to study the condition of the colon. Peutz-Jeghers polyps resemble adenomas in appearance, can reach several centimeters and have a stalk. Sometimes hyperemia and erosion of the epithelium are noted. - Histological analysis
. Cytomorphological study of biopsy specimens is necessary for differentiation from other neoplasms. The disease is characterized by preservation of all layers of the mucous membrane, absence of cellular atypia and pathological mitoses, proliferation of stroma and smooth muscle fibers. According to their tissue structure, polyps are hamartomas.
A general blood test reveals signs of anemic syndrome - a decrease in the amount of hemoglobin and the number of red blood cells. A biochemical blood test reveals a decrease in the level of total protein. All patients undergo a coprogram; to confirm bleeding from the gastrointestinal tract, the Gregersen test for occult blood is performed. According to indications, a cytological analysis of skin biopsies from pigmented areas is performed.
Differential diagnosis for the Peutz-Jeghers symptom complex is primarily carried out with other forms of polyposis (diffuse familial, juvenile). The main diagnostic differences are the presence of hyperpigmentation of individual areas of the skin and the specific histological structure of polyps. It is also necessary to differentiate the disease from lentigo multiplex. To examine a patient with signs of polyposis syndrome, a specialist gastroenterologist, proctologist, and dermatologist are involved.
Standard Treatments
Patients should first consult with a clinical geneticist or genetic counselor. Because there is no cure for Peutz-Jeghers syndrome, treatment mainly focuses on observation and control of symptoms. After initial diagnosis, people over 8 years of age or who have symptoms are advised to undergo endoscopic and small bowel testing. The latter can be done with magnetic resonance imaging of the intestines (magnetic resonance enterography, MR enterography) or by swallowing a capsule that takes internal images of the gastrointestinal tract (video capsule endoscopy). Gynecological and breast examinations are also recommended for women over 18 years of age. Testicular examination is recommended for men.
After the initial evaluation following diagnosis, endoscopy, colonoscopy, and small bowel screening should be performed every 2 to 3 years to identify polyps and potential tumors. Women are recommended to have an annual mammogram. For men, testicular ultrasound can be done once every two years.
Because Peutz-Jeghers syndrome increases the risk of breast, uterine, and ovarian cancer, affected women may undergo prophylactic mastectomy, hysterectomy, or salpingo-oophorectomy (surgical removal of the breast, uterus, fallopian tubes, and ovaries, respectively).
Polyps larger than 1 cm are removed endoscopically to avoid polyp-related complications such as bleeding and intussusception. These complications may require surgery. If a patient is undergoing surgery, endoscopic removal of polyps (polypectomy) is performed at the same time as surgery to reduce the risk of recurrent complications and surgery.
In cases where dark pigment spots (melanocytic spots) have an extremely negative psychological impact on affected people, they can be partially removed using laser therapy.
Treatment
Despite the fact that Peutz described Peutz-Jeghers-Touraine syndrome back in 1921, the symptoms and treatment of this disease have not yet been sufficiently studied. At the moment, a comprehensive treatment for this disease has not been developed. Since hundreds of polyps grow in the intestines during the syndrome, it is impossible to remove all of them prophylactically due to the high risk of such an operation. Therefore, patients with Peutz-Jeghers syndrome undergo many surgical interventions throughout their lives to remove polyps when they reach critical sizes.
Paraoncological dermatoses
PARAONCOLOGICAL DERMATOSES
Paraoncological dermatoses (syn.: paraneoplastic dermatoses) are skin reactions to malignant neoplasms of internal organs and systems that change the physiological processes in the body and lead to the accumulation in it of usually biologically active substances (hormones, growth factors, etc.) and tumor antigens that promote emergence of new clinical symptoms.
Lesions of the skin and mucous membranes can be divided into three groups:
- hereditary syndromes in which a malignant tumor is combined with diseases of the skin and mucous membranes;
- paraneoplastic dermatoses caused by the influence of the tumor on the metabolism, immunity and regulatory systems of the body;
- metastasis of malignant tumors to the skin and mucous membranes.
Hereditary syndromes Gardner syndrome
Gardner syndrome is a hereditary symptom complex that includes epidermoid cysts, fibromas, osteomas in combination with rectal polyposis.
Type of inheritance: autosomal dominant.
Predominant age. Develops at the age of 4-10 years, rarely later.
Clinical picture
- Epidermoid cysts on the skin of the face, scalp, limbs, and less commonly the chest.
- Osteomas develop mainly in the jaw and sphenoid bones.
- Polyposis of the colon and rectum, but polyps are often found in the small intestine and stomach. Polyps in the large intestine tend to become malignant.
Diagnosis is based on clinical data and special methods of examining the digestive tract.
Treatment. Surgical removal of polyps.
Associated malignancies: cancer of the gastrointestinal tract, as well as bone and endocrine glands.
Cowden syndrome
Cowden syndrome is a rare hereditary symptom complex. It is characterized by multiple hamartomas of ecto- and mesodermal origin in combination with a predisposition to malignant tumors, especially of the mammary and thyroid glands. The syndrome is named after the family in which such patients were first noted.
Predominant age. 4-75 years (half of the patients are under 40 years old); men get sick more often.
Type of inheritance: autosomal dominant with variable gene expressivity.
Clinical picture. A constant symptom of the disease, varying only in its intensity, is mucocutaneous wart-papillomatous lesions. Initially, multiple, skin-colored lichenoid papules (up to 4 mm in diameter) appear on the eyelids, around the eyes and mouth. Subsequently, they merge and form surfaces in these areas that resemble cobblestones. The process can involve the lips, mucous membrane of the cheeks, pharynx, and larynx. On the palms and soles, extending to the fingers, there are small translucent keratotic layers. Angiomas, lipomas, and melanoma are also possible.
General state. Skin changes are usually combined with fibrous cystosis or fibroadenoma of the mammary glands in women with a tendency to malignancy (30% of cases) and goiter, adenoma or thyroid cancer. Other anomalies are less common (gothic palate, skin dyschromia, skeletal disorders, polyposis of the digestive tract, gynecological pathology, primary T-cell immunodeficiency).
Diagnosis is based on clinical and histological data.
Differential diagnosis
- with Darier's disease;
- tuberous sclerosis.
Treatment
Surgical removal of tumors, clinical observation with periodic mammography and monitoring of the thyroid gland. Avoid the use of estrogens.
Course and prognosis. New tumors can appear throughout life.
Synonym: multiple hamartoma and neoplasia syndrome.
Associated malignancy: breast adenocarcinoma.
Neurofibromatosis (see p. 115)
Associated malignancy:
- neurofibrosarcoma;
- pheochromocytoma;
- rhabdomyosarcoma, etc.
Peutz-Touraine-Jeghers syndrome
The main symptom of this hereditary syndrome is small multiple pigment spots (lentigo) on the lips and mucous membranes of the mouth. Polyposis of the stomach, small and large intestines. Polyps are hamartomas and are found in most patients.
Type of inheritance: autosomal dominant.
Floor
•Lentigo is found in infants and young children./>
•Polyps - in older children and adults under 30 years of age. Men and women get sick equally often./>
Patients are predisposed to breast, ovarian and pancreatic cancer.
Flow. Lentigo occurs already at birth or appears in infancy and early childhood.
Clinical picture. On the skin of the lips, mucous cheeks, face, around natural openings, palms, soles, spots appear, round and oval in shape, 2-5 mm in size, dark brown or black. The rashes on the face are smaller than on the palms, soles and mucous membranes. Spots on the lips may disappear over time, but the pigmentation of the mucous membrane remains for life. There are polyps of the small intestine, and less often - of the large intestine. Intussusception and intestinal obstruction are possible.
Diagnostics
- Anamnesis.
- Clinical picture.
- Detection of gastrointestinal polyps.
treatment has been developed. Surgical removal of bleeding polyps or those exceeding 1.5 cm in size is performed. Synonyms
- Periorificial lentigiosis.
- Peutz-Jeghers syndrome.
• Polyposis pigmented-spotted. Associated malignancy:/>
- mammary cancer;
- ovarian cancer;
- pancreas cancer.
Paraneoplastic dermatoses are obligate (always combined with malignant
tumor)
Acanthosis nigricans
Associated malignancy:
- gastric adenocarcinoma;
- uterine cancer;
- ovaries;
- prostate gland;
•lungs;
• lymphoreticular system. Acrokeratosis psoriasiformis Bazexa />
Acrokeratosis psoriasiformis Bazexa is a para-oncological dermatosis.
Floor. More common in men.
Clinical picture. It is characterized by erythematosquamous lesions with acral localization. Skin changes
with peeling on the skin of the nose, ears, hands, feet. Gradually the rash becomes more generalized. Nail dystrophy and paronychia occur. Changes in facial skin may be similar to manifestations of eczema or lupus erythematosus, while acral lesions (on the hands, feet) are more reminiscent of psoriasis.
Histologically , hyperkeratosis, parakeratosis, focal spongiosis and inflammatory mixed infiltrate in the dermis are revealed.
Diagnosis is made on the basis of clinical and histological data.
Differential diagnosis is carried out with seborrheic eczema, allergic dermatitis, Reiter's syndrome, psoriasis, lupus erythematosus.
Treatment: identification and radical removal of a malignant neoplasm, oral etretinate (0.5 mg/kg per day).
Associated malignancy:
• cancer of the upper respiratory tract;
- digestive tract.
Carcinoid syndrome
Sudden temporary dark red or purple erythema resulting from blood flow to the face and neck; diarrhea, cramping abdominal pain, right ventricular failure, asthma, edema, telangiectasia, cyanosis, pellagra-like rashes, signs of Cushing's syndrome. In the urine - an increase in the level of the serotonin metabolite 5-hydroxy-indoleacetic acid.
Associated malignancy: carcinoid tumor in the digestive tract, most often in the small intestine, lung, liver metastases
Erythema garlandum migrans Gammel
Erythema garlandum migrans of Hammel is an obligate para-oncological dermatosis, the development of which is associated with the autoimmune mechanism of action of tumor tissues. Described by Gammel in 1952. It is often observed several months (sometimes 2-4 years) before clinical signs of malignant neoplasm of internal organs.
Clinical picture. Skin lesions are characterized by multiple spotty, sometimes somewhat swollen, rash, the first elements of which usually appear on the face, and then spread to the skin of the trunk, proximal limbs in the form of bizarre figures, rings, garlands, quickly moving across the skin (within several hours or days ). Fine-plate peeling and mild itching are possible. The palms and soles are not affected. Clinically and histologically, the disease resembles Darier's annular erythema.
Diagnosis is based on clinical data. A thorough oncological examination of the patient is necessary.
Treatment: radical removal of a malignant neoplasm of internal organs leads to complete regression of skin lesions.
Associated malignancy:
- lungs' cancer;
- mammary glands;
- genitals;
- stomach.
Necrolytic erythema
Necrolytic migratory erythema is caused by the development of glucagonoma, a tumor growing from the e-cells of the pancreatic islets. The tumor produces glucagon and is accompanied by very characteristic rashes.
Etiology. Most cases of necrolytic migratory erythema are caused by excess glucagon in the blood, but its pathogenesis is unknown.
Age. Middle and old.
Clinical picture. Inflammatory plaques with peripheral growth and epithelialization in the center. The plaques merge, arranged in the form of rings and arcs, which gives the lesions the appearance of a geographical map, along the edges of which bubbles, crusts and scales form. The rashes are located on the flexor surface of the limbs, in large folds, around natural openings. The tips of the fingers are red, shiny, with erosions.
Research methods. Increased fasting plasma glucagon level more than 1000 ng/l.
Diagnostics
- Clinical picture.
- Biopsy results.
- Determination of glucagon level in the blood.
Differential diagnosis
- Enteropathic acrodermatitis.
- Exudative psoriasis.
- Candidiasis.
- Benign familial pemphigus Gougereau-Hailey-Hailey.
Treatment. Surgical removal of the tumor.
Associated malignancy:
- glucagonoma;
- severe liver cirrhosis;
- lung cancer.
Paget's disease of the breast
Paget's disease of the breast is intraductal breast cancer. It may be limited to damage to the epidermis of the breast or combined with breast carcinoma, with the epidermis being affected secondarily.
Age, gender It occurs mainly in women 50-60 years old.
Clinical picture. The disease is characterized by a focus of congestive hyperemia with clear boundaries in the area of the areola and nipple of the mammary gland. Signs of eczematization may be observed on the surface of the lesion: peeling, erosion, weeping, serous and hemorrhagic crusts, itching. Protrusions from the nipple of a serous or hemorrhagic nature are possible. Gradually, the lesion becomes denser, covers the skin of the chest, and its borders rise. The nipple retracts or disappears, and an area of compaction is determined in its depth; Regional axillary lymph nodes may enlarge. The course is steadily progressing. Primary breast cancer spreads downward to the epithelium of the lobules, as well as upward and outward to the epidermis, where skin lesions develop. In the late stage of the disease, cancer cells penetrate the wall of the duct or lobules and infiltrate the connective tissue of the mammary gland - lymphatic metastases develop.
The diagnosis is made based on the clinical picture, the course of the process, and the results of histological examination.
Differential diagnosis with
- eczema of the nipples;
- Bowen's disease;
- superficial form of basalioma;
- melanoma.
Treatment: surgical removal of the tumor.
Associated malignancy: intraductal adenocarcinoma of the breast.
Optional ( often combined with a malignant tumor)
Paget's disease extramammary
Extramammary Paget's disease is a malignant tumor that affects the perianal region, external genitalia and armpits.
Age, gender It occurs both in women (more often) and in middle-aged and elderly men.
The clinical picture is characterized by red spots or plaques with uneven outlines, a macerated surface, cortical layers, and itching. Often tends to grow invasively and transform into squamous cell carcinoma. Often associated with malignant neoplasms of internal organs. Histologically, it does not differ from the mammary localization of Paget's disease, but the extent of the pathological process significantly exceeds the visible size of the lesion, which causes frequent relapses after treatment.
Diagnosis is based on clinical and histological findings.
Differential diagnosis with
•Bowen's disease;
- pagetoid epithelioma;
- eczema.
Lead tactics. Be sure to examine the rectum, cervix and urethra in search of the primary tumor.
Treatment
- Surgical excision with a wide coverage of apparently unchanged skin.
- Prescribe bleomycin 15 mg IM daily; per course 5-6 mg/kg, intervals between courses 1.5-2 months, or prospidin IM or IV 0.2 g/day; per course 3-4 g, and in severe cases 5-6 g.
- For superficial forms, localized on the genitals or in the perineum, external chemotherapy is performed: 5% fluorouracil, fluorofuric, 30-50% prospidine, 0.5% colchamine ointment.
- Cryodestruction and laser therapy are also used.
Associated malignancy:
- uterine cancer;
- ovarian cancer;
- prostate cancer;
- rectal cancer.
Bowen's disease
Bowen's disease is an intraepidermal skin cancer that necessarily transforms into squamous cell carcinoma.
Age, gender It develops more often in older people (70-80 years old) of both sexes.
Etiology and pathogenesis. The development of the disease is caused by the action of UV irradiation, long-term trauma to the skin, and when the process is localized in closed areas of the skin - contact with arsenic salts (medicines, industrial production, etc.). The lesion is most often located on the torso, upper extremities, in the perineum, and sometimes on the mucous membrane of the mouth.
Clinical picture. The process is represented by a solitary dense plaque of irregular or round shape, covered with white or yellowish scales, which are easily removed with the formation of erosions and weeping, but without signs of bleeding. Erosions can be covered with serous and hemorrhagic crusts. The size of the lesion is from 2 mm to the child’s palm. Important features of Bowen's disease include uneven growth of the lesion along the periphery, its diversity due to areas of atrophy, erosion, hyperkeratosis, warty growths, and elevation of the marginal zone.
Course and prognosis. The course is slow and steadily progressing. Squamous cell carcinoma can develop even in the early stages of the disease. Relapses occur as a result of inadequate treatment.
The diagnosis is made based on the clinical picture and the results of histological examination.
Differential diagnosis is carried out with eczema, psoriasis, warty skin tuberculosis, squamous cell and metatypical cancer, solar keratosis, senile keratoma, basal cell carcinoma, bowenoid papulosis.
Treatment: for small lesion sizes (up to 2 cm), applications of 30-50% prospidine and 5% fluorouracil ointments are used; for lesion diameters greater than 2 cm, cryodestruction, surgical and electrosurgical treatment are indicated; removal with carbon dioxide laser. If the lesion is localized on the mucous membrane, 5% fluorouracil ointment is prescribed, and aromatic retinoids are prescribed orally (tigazone at the rate of 1 mg/kg per day for 1-2 months). X-ray therapy is ineffective.
Associated malignancy:
- gastric adenocarcinoma;
- uterine cancer;
- ovaries;
- prostate gland;
- lungs;
- lymphoreticular system. D
Exfoliative dermatitis
Subacute generalized exfoliative dermatitis is a specific erythroderma, which is a manifestation of T-cell lymphoma of the skin. The development of the disease is facilitated by impaired immunity.
Clinical picture. Most often, erythroderma is preceded by the appearance of lesions resembling eczema or neurodermatitis. The resulting clinical picture is characterized by generalized erythema covering the entire skin, swelling of the skin, profuse large-plate peeling, as well as palmoplantar keratosis, hair and nail loss, itching, chills, and enlarged peripheral lymph nodes. In the blood - leukocytosis, monocytosis, eosinophilia and increased ESR.
Synonym: Wilson-Brock erythroderma.
See Lymphomas of the skin.
Associated malignancy: lymphoproliferative diseases.
Itchy skin
Associated malignancy:
- lymphoma;
- leukemia
Probable (sometimes combined with a malignant tumor) Acquired ichthyosis Associated malignant neoplasm:
- lymphogranulomatosis;
- testicular sarcoma.
Pemphigoid bullous
Associated malignant neoplasm: malignant tumors of various types and locations. .
Dühring's dermatitis herpetiformis
Associated malignancy:
- gastric adenocarcinoma;
- breast cancer, etc.
Erythema annular centrifugal Daria
Associated malignancy:
- gastric adenocarcinoma;
- mammary cancer;
- lung cancer.
Skin metastases
Metastases of malignant tumors are single or multiple nodes localized in the skin or subcutaneous tissue. According to the mechanism of occurrence, hematogenous, lymphogenous and implantation metastases are distinguished.
Frequency. Skin metastases are found in 0.7-9% of all patients with malignant tumors.
Predominant age. Anyone, but more often the elderly.
Course and prognosis. The prognosis is usually unfavorable. The average life expectancy of patients after detection of skin metastases is only a few months.
Elements of the rash. A node, a fibrous plaque, of dense consistency, protruding above the surface of the skin. Metastases are usually detected when their size exceeds 5 mm. The color of the rash is pink or red. Melanoma metastases appear as blue, gray or black intradermal nodules.
Types of skin lesions
Mammary cancer. With the lymphogenous spread of the tumor, inflamed hyperemic plaques form on the skin of the affected gland, as in erysipelas, dense flattened papules and plaques, telangiectasia or nodes.
Colon cancer. Metastasizes to the skin of the abdomen, perineum, scalp and face. Metastases are represented by inflammatory plaques, less often nodes on the stalk or broad base on the buttocks; groups of richly vascularized formations in the groin area and on the scrotum.
Lung cancer. More often it gives metastases in the form of reddish nodes located symmetrically on the scalp or torso. The localization of the rash may coincide with the course of the intercostal vessels.
Kidney cancer. Metastases can be single or multiple, usually in the form of vascular formations similar to telangiectatic granuloma, sometimes they are located on a stalk.
Predominant localization is on the head and neck, less often on the trunk and limbs.
Melanoma. Melanoma metastases are represented by single or multiple dark-colored nodes, but sometimes there are also non-pigmented forms. Metastases and relapses of melanoma can appear both in distant areas of the skin and in nearby ones.
Bladder cancer and ovarian cancer. These tumors can spread to the skin of the abdomen and groin area, forming inflammatory, hyperemic plaques that resemble erysipelas.
Diagnostics
- A history of a malignant tumor of internal organs.
- Histological examination.
Treatment. If the patient's condition allows, excision of single metastases.
Cancer risk
Patients with Peutz-Jeghers syndrome have a significantly higher risk of developing gastrointestinal cancer than healthy people. Most often, oncology affects the large intestine, small intestine and pancreas. Women often experience breast cancer (45% of cases). It is precisely in the high probability of developing cancer that the danger of the disease lies; Peutz-Jeghers syndrome must be controlled using several clinical procedures.
Given the increased risk of developing cancer, all patients should undergo x-rays of the small intestine approximately once every 2 years.
Gastroscopy and colonoscopy will help the attending physician monitor the condition of the stomach and intestines, as well as monitor the size of polyps. Even with successful surgery to remove a polyp, the likelihood of recurrence is high, which is why these examinations are absolutely necessary.
Women over 25 years old should have a mammogram every year to detect cancer at an early stage.
In the diagnosis of cancer, testing for the presence of carcinoembryonic antigen in the blood plays an important role. This is a chemical produced by colon, breast or lung cancer.
It is also important to determine other cancer antigens (also known as tumor antigens) - substances produced in cancer-affected cells of any organ. Their nature depends on the location of the tumor, which is why these substances play an important role in diagnosis.
Most patients with Peutz-Egers syndrome do not live to age 60, dying from cancer of the pancreas (11%), stomach (57%), intestines (85%), and breast (45%). The risk of developing cancer of the lungs, testicles, cervix, and ovaries also increases slightly. If cancer is detected at a late stage, it is fatal. This is why timely diagnosis is so important.
results
In some genetically determined syndromes, malignant tumors are an integral part, an example is FAP (OMIM#175100). This syndrome has an autosomal dominant pattern of inheritance with a prevalence of 1 case per 10,000 births. It is characterized by the appearance of many polyps, the malignancy of which occurs in 100% of cases, and it is with this syndrome that it is possible to identify affected individuals before the appearance of cancer. The clinical picture of the disease includes three phenotypes, among which the classic form of the syndrome has severe and moderately severe forms. Severe (classic) form of FAP: more than 2000, 5000 polyps (or profuse polyposis) in the left half of the intestine, early appearance of polyps, rapid malignancy. Moderate (classic) form: the presence of hundreds or more adenomatous polyps, typically also localized in the distal parts of the colon. The third, mild form of FAP, also called “attenuated” or ACAP syndrome: a small number (more than 20, but less than 100) of adenomas, in most cases in the right side of the colon, and appears at a later age (over 15 years). In 10% of carriers of the first two forms of the syndrome, polyps appear before the age of 10 years, and by the age of 20, polyps develop in 95% of patients, most of them have a family history. Adenomatous polyps are benign, but some of them, if not surgically removed, transform into colorectal cancer by an average of 35 years of age [1].
According to our data, the earliest diagnosis of malignancy was in one of the patients with a familial form of FAP, in whom polyps were diagnosed at the age of 4 years, and a colonectomy was performed at the age of 14 years. Cancer of the remaining part of the rectum, identified during observation, was surgically removed at an early stage at the age of 21. In Fig. 1 picture of the classic form of FAP syndrome, in Fig. Figure 2 (a, b) shows the histological and cytological picture of an adenoma in FAP syndrome.
Rice. 1. Diffuse polyposis of the colon.
Rice. 2. Tubulovillous adenoma in FAP syndrome (a), histological staining with hematoxylin and eosin, ×100; cytological preparation (b).
FAP syndrome is a multitumor syndrome in which patients are predisposed to the appearance of benign and malignant formations of mesenchymal, ectodermal and endodermal origin in different organs. For example, congenital hypertrophy of the retinal pigment epithelium (CRPE) of the eye, epidermoid cysts, abnormalities of bones and teeth, and osteomas are not associated with malignancy. Desmoid and adrenocortical tumors, polyps of the duodenum and stomach, and liver adenomas carry malignant potential. Tumors of the thyroid gland, ovaries, pancreaticobiliary zone, brain and liver in this syndrome often manifest as malignant [2-6].
In the third, mild form of FAP, extraintestinal manifestations are rare, and the risk of developing colon cancer depends on the severity of polyp damage. Polyps, as the most permanent component of the syndrome in patients with FAP, can occur in the stomach and duodenum. Formations such as skin fibromas and epidermoid cysts, lipomas, multiple osteomas of the facial bones, most often the lower jaw, dental growth abnormalities, juvenile nasopharyngeal angiofibromas do not require active treatment. However, desmoid tumors can be clinically aggressive, manifesting as one or more formations in the abdominal cavity or along the spine or extremities. Their joint manifestation with polyps of the gastrointestinal tract, osteomas of the facial bones, and skin fibromas is referred to as Gardner syndrome (a variant of FAP), in which the risk of developing cancer, including the stomach, is increased.
In our patients with Gardner's syndrome, osteomas of the facial bones, intestinal polyps and polyps in the stomach were diagnosed at 12 and 16 years of age. Moreover, one of them was diagnosed with stomach cancer at the age of 23 years. In the families of these patients there were indications of gastrointestinal tumors in 1st and 2nd degree relatives, from which they died in old age (over 54 years).
For patients with FAP, as well as for relatives, there is a high risk of developing liver tumors, including hepatoblastoma or hepatocellular cancer, which can occur earlier than polyps (at 2-3 years of age). However, despite the high risk for FAP patients, these tumors are rare [7, 8]. In addition, there is a risk of brain tumors, which also occur at an early age, before polyps appear. Histologically, this may be medulloblastoma (80%), pineoblastoma, astrocytoma or adenoma (pinealoma), or pineal cysts. It is important to note that a carrier of the syndrome may develop a brain and liver tumor together. The combination of multiple adenomas in the colon with a brain tumor is known as glioma polyposis syndrome or Turcotte syndrome, which is also classified as a variant of FAP.
Interestingly, patients with FAP have ocular GPES, which is often associated with thyroid cancer [8]. TC is a common component of FAP and affects from 2 to 11.8% of patients. Histologically, it is usually papillary cancer, but in 1/3 of cases it has a cribriform structure, which is almost never found in the general population (0.2%), occurs at a young age (20 years) and has a favorable prognosis. Benign thyroid nodules occur in 38–79% of patients [9].
In our patient with FAP, colon adenomas were diagnosed at 7 years of age; they were removed endoscopically as they were diagnosed; subtotal colectomy with ascendorectal anastomosis was performed at 14 years of age. At the age of 16, nodules were discovered in her thyroid gland, and at the age of 17, thyroid cancer was removed at an early stage. Histologically, it is a typical differentiated papillary carcinoma (Fig. 3). The father of the patient, also with FAP syndrome, died from progression of colon cancer at the age of 40.
Rice. 3. Papillary thyroid cancer of a patient with FAP syndrome. Hematoxylin and eosin staining. ×40, ×100.
In another patient, thyroid cancer, diagnosed at the age of 19 (histologically follicular), became the first manifestation of a malignant phenotype (Fig. 4). In addition, she was diagnosed with HPES. Hemicolectomy was performed at the age of 27; moderately differentiated adenocarcinoma and multiple colon polyps with signs of malignancy were removed.
Rice. 4. Follicular thyroid cancer of a patient with FAP syndrome. Hematoxylin and eosin staining. ×40.
For this syndrome, compared to the general population, the female/male ratio (17:1) is much higher (3:1). TC is diagnosed first in relation to the identification of the syndrome in a third of patients with FAP, and this is the reason for searching for adenomas in the colon in individuals with early development of thyroid cancer. It is also important that patients with FAP may experience cosegregation of brain tumors and papillary thyroid cancer [10]. An interesting observation was that patients with FAP associated with brain or liver tumors, or thyroid cancer, had HPPES, and relatives from their families had the same tumors or intestinal polyps or HPPES [11]. Thus, the diagnosis of HPPES in a patient with FAP is a sign that can be associated with the appearance of malignancy not only in the thyroid gland, but also in other organs. Desmoid tumors are found in families of patients with FAP in 25% of first-degree relatives and even in 8% of third-degree relatives, which can also be an additional sign in identifying the syndrome.
Molecular genetic basis of FAP syndrome. Predisposition to FAP is caused by a germline mutation in the APC (adenomatous polyposis coli) gene, which was identified on chromosome 5q21 in 1991 [12, 13]. The APC gene is a suppressor gene and consists of 15 exons (exon 15 occupies ¾ of the coding sequence). The APC gene encodes a protein with a molecular weight of 311.8 kDa and is the main regulator in the Wnt signaling pathway. As part of a protein complex, the APC gene is involved in regulating the phosphorylation and degradation of β-catenin. APC protein plays a key role in the adhesion of epithelial cells through binding to E-cadherin and microtubules, regulates cell migration in intestinal crypts, proliferation, and is involved in apoptosis and intracellular signal transmission. In addition, APC is a promoter of chromosome stability [14, 15]. Most mutations in the APC gene are large deletions that are difficult to detect. Most often, germline mutations include intragenic insertions, deletions causing a reading frame shift, point mutations leading to premature termination of protein synthesis and functional inactivation of the APC protein. To date, more than 900 mutations are known. As for patients with a mild form of FAP, some of them have mutations in the APC gene or in the MUTYH gene, the product of which is involved in DNA excision repair, and biallelic mutations in this gene lead to the development of FAP. If a mutation is not detected in any of these genes, one of the reasons may be a large deletion leading to loss of the gene. It is interesting to note that in patients with FAP, loss of APC gene function begins with germline loss of one allele of the gene; the mutation of the second allele depends on the location of the first mutation. It has been shown that in carriers of mutations in codons 1194-1392, the second copy of the gene is more often lost through large deletions. Mutations outside this region are most often point mutations, which lead to premature synthesis of the APC protein. However, the closer the mutation is located to codon 1300, the higher the probability of deletions and complete shutdown of the second copy of the gene, which entails a selective advantage acquired by the cell [16]. Therefore, the microscopic polyps in the carrier of these mutations are not identical and grow at different rates. In addition, analysis of early adenomas (3 mm) from patients with FAP with known mutations in the APC gene showed that further development of tumorigenesis requires additional combined effects of various environmental factors, modifying genes, acquisition of mutations in the K-ras, TP53 genes, deletions of chromosome 18q and chromosome 1p, affecting different pathways of gene function and leading to an increase in chromosomal instability [17].
Germinal mutations in the APC gene cause a wide range of clinical manifestations of the disease, which manifest themselves at different ages throughout life, and the severity of the syndrome depends on the type of mutation in the APC gene. The table shows data from different authors on the localization of mutations in the APC gene and the possible phenotypic manifestations of the disease associated with them (see table).
Types of mutations in the APC gene and the severity of the disease
As can be seen from the table, despite the not very clear boundaries of the intervals of the APC gene between the severe and classic forms of manifestation of the syndrome, the mutations identified in the gene can serve as a guide to the possible severity and variant of the manifestation of the phenotype. For the attenuated form of FAP, the boundaries of the intervals in the APC gene have been established. The milder course of the disease in this form of FAP is explained by the preserved expression of the alternatively spliced transcript of the APC gene, which does not contain a mutation, since it does not contain exon 9 [21, 29]. Based on the obtained correlations, it is possible to assess the risk of cancer development and plan treatment for patients with FAP. Thus, a germline mutation identified in one of the codons 1250–1464 suggests more aggressive treatment methods, especially the mutation in codon 1309, which is associated with the early development of colon cancer [19].
It is interesting to note that germline mutations in patients with FAP and embryonal liver cancer (hepatoblastoma) are mostly localized at the 5' end of codon 1230 or codon 1061, and when a patient with FAP had a brain tumor and liver cancer combined, the mutation in the APC gene affected that the same region as in hepatoblastoma [8,11]. In addition, it has been shown that the risk of thyroid cancer is mainly due to a germline mutation at the 5' end of exon 15, which is considered a “hot spot” of the APC gene [8]. However, testing of this gene in patients with FAP and thyroid cancer identified mutations in exons 4, 8 and 9 and it was shown that they can occur throughout the gene [30, 32].
Clinical diagnosis of neoplasia. The risk of inheriting a germline mutation by offspring with this syndrome is 50%. A germline mutation is found in 80% of patients with polyposis, and in 30% of them it occurs de novo or as a result of mosaicism. Genetic testing begins with a family member who is a carrier of the syndrome. Other relatives in the family are tested after a mutation in the APC gene is identified in a carrier of the syndrome. If a mutation in a patient affected by polyps is not detected in the APC gene and in the MUTYH gene, 1st degree relatives are examined in the same way as patients with FAP. If a mutation is detected, all relatives suspected of having FAP undergo direct DNA testing, and monitoring includes annual colonoscopy with polyp biopsies, starting at age 10–12, every 2 years until age 35. For relatives, colonoscopy should begin at puberty or based on symptoms such as diarrhea, abdominal pain, blood in the stool, metabolic disorders (decreased protein levels, cholesterol, hypokalemia), dysbiosis, secondary immunodeficiency. For older patients, a colonoscopy is performed at the first examination, then sigmoidoscopy every 1-2 years. It is recommended to start endoscopy of the stomach and small intestine at the age of 25-30, depending on the clinical manifestations found, and repeat every 2-3 years until the age of 50. According to the literature, in patients after prophylactic colonectomy, duodenal cancer is the leading cause of death; the earliest age at diagnosis of this cancer is 17 years [33]. Polyps in the fundus of the stomach are also prone to malignancy. If there are more polyps in the patient's intestine than can be removed, colonectomy with ileorectal anastomosis or ileoproctocolonectomy followed by ileoanal anastomosis and observation of the condition of the rectum is recommended [34].
In families where there are one or two family members with a mild form of FAP, testing begins in later adolescence (after 15 years).
Given the high risk of developing hepatoblastoma in children, analysis of the level of α-fetoprotein and ultrasound examination of the abdominal cavity should begin before 2 years of age, repeat every six months until 6 years of age, for some the risk remains until 15 years of age. Brain examinations begin after 2 years of age. It should be remembered that the above tumors can occur before polyps appear in the intestines. Indications of hepatoblastoma in one of the family members are the reason for starting examination of the patient at 0.6 years of age with DNA diagnosis for carriage of mutations in the APC gene. Palpation examination of the thyroid gland annually, starting from the age of 15 years, and an ultrasound scan of the thyroid gland every 3 years. If necessary, a fine-needle biopsy is used. It should be remembered that in some patients and their relatives all of the above tumors may appear together. A thorough annual thyroid examination is also recommended when a patient or a family member is diagnosed with HPES or a germline mutation in the APC gene is detected at codons 463−1387.
Early diagnosis and treatment planning of patients with FAP can be helped by identifying not only extraintestinal benign and malignant manifestations and associations between them in this disease, but also genotype-phenotype correlations in carriers of mutations in the APC gene.
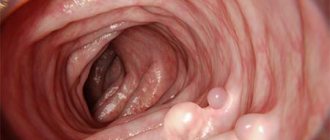
Peutz-Jeghers syndrome (OMIM#175200) refers to the syndromes of multiple hamartomatous polyposis. It has an autosomal dominant type of inheritance, with a prevalence of 1 case in 50,000 and 1 in 200,000 newborns [35]. A hamartoma is a nodular formation that occurs as a result of a violation of the embryonic development of organs and tissues, consisting of the same components as the organ where it is located, but differs in the degree of differentiation. The clinical picture of the disease includes one of the main characteristics of the syndrome - polyps, which can be located in any part of the digestive tract. Polyps from 1 to 100 (size from 0.1 to 3 cm in diameter), usually benign, develop in 90% of patients, and in 1/3 of patients are detected by 10-13 years. In addition, they can appear in the nose, bronchi, organs of the reproductive system, kidneys, urethra, urinary and gall bladder.
In the intestine, polyps upon histological examination consist of normal cellular elements of the digestive tract, but with altered architecture caused by the elongation of the plate of the epithelial component of the intestinal mucosa into the stroma of the polyp and the branching of smooth muscle fibers, which creates a picture of epithelial invasion into the thickness of the intestinal wall [36, 37]. The second characteristic of the syndrome is melanin pigmentation (from 1 to 5 mm in diameter) of brown or light brown color at the border of the skin and mucous membranes (typically lips and cheeks), perianal area, on the palms and soles, and intestinal mucosa. Hyperpigmentation occurs at birth, can appear at the site of injury, inflammation and fade or disappear by puberty or with age (Fig. 5).
Rice. 5. Pigmentation of the mucous membrane of the lips and oral cavity in Peutz-Jeghers syndrome.
Hamartomatic polyposis in the intestinal tract can be accompanied by severe complications: ulceration, bleeding, causing intussusception, obstruction (usually of the small intestine) and even necrosis, which are detected in 33% of carriers of the syndrome in early childhood (up to 10 years), and by the age of 20 - in 50% [38]. It is now known that for patients with Peutz-Jeghers syndrome, the risk of developing cancer of any location is 15 times higher than in the general population and by the age of 65 years of life is 93% [39]. The most common sites of cancer occurrence are the gastrointestinal tract and breast. Most often, cancer occurs in the small intestine (96%), colon (27%), stomach (24%), and rectum (24%) [40]. The average age of diagnosis of gastric cancer is 30 years, but can occur at 10 and 20 years of age [39]. The risk of breast cancer (BC) is similar to that of patients with BRCA1 or BRCA2 mutations (cumulative risk 45%). Bilateral lesions are characteristic; the earliest age of the patient at diagnosis of breast cancer is 19 years [41]. 75% of women develop fibroadenomas, cysts in the mammary glands, uterine leiomyomas (44%) with a high risk of malignancy.
In addition, benign diseases of the thyroid gland, including multinodular goiter, are detected in 50-70% of carriers of the syndrome, and from previous follicular adenomas, follicular thyroid cancer occurs in 5-10% [39].
Patients with Peutz-Jeghers syndrome from a young age have a risk of developing cancer of the endometrium, cervix, ovaries, urethra, testicles, which is often bilateral and multifocal, esophagus, lungs, and pancreas. On average, the risk of cancer appears at 42 years of age [42, 43].
In our study, a patient aged 38 years underwent subtotal resection of the colon for primary multiple synchronous cancer. On the skin in the area of the lips and mucous membranes of the cheeks and perianal area, pigmentation characteristic of this syndrome was detected. Indications of constant diarrhea in his 7-year-old son led to an examination and identification of small polyps in the jejunum and polycystic kidney disease. The removed polyps were tubular adenomas upon histological examination (Fig. 6). Other manifestations of the syndrome in the child include isolated spots of hyperpigmentation on the fingers. Subsequently, in order to prevent complications in the child, endoscopic removal of polyps was performed repeatedly.
Rice. 6. Tubular adenoma. Hematoxylin and eosin staining. ×75.
Clinical diagnosis of the syndrome is based on identifying one of the following manifestations of the disease:
1) 2 or more histologically confirmed hamartomatous polyps;
2) any number of polyps identified in a patient who has a close relative who is a carrier of the syndrome;
3) hyperpigmentation in the patient in places typical for the syndrome and the presence of a relative in the family who is a carrier of this syndrome;
4) any number of polyps in combination with the presence of hyperpigmentation in typical places in one individual [39].
Molecular genetic basis of Peutz-Jeghers syndrome
The cause of the syndrome is a germline mutation in the STK11 gene, which encodes the serine/threonine kinase protein. The STK11 gene is a suppressor gene, mapped on chromosome 19p13.3, includes 9 exons, encodes a protein consisting of 433 amino acid residues [44, 45]. The main types of mutations are small deletions/insertions, nonsense, missense, or large deletions, leading to premature termination of protein synthesis [46]. The function of the STK11 gene is complex, but not fully established. However, it is known that it regulates cell proliferation by arresting the G1 cell cycle, regulates the Wnt signaling pathway by interacting with the p53 protein, and is involved in apoptosis [47, 48]. The STK11 gene has an important role in the orientation of cells within a tissue, as it affects cell polarity and is involved in intermembrane protein interactions [49, 50]. The STK11 protein is involved in the inhibition of the mTOR (mammalian target of rapamycin) pathway, which is also known as the rapamycin-associated protein complex, acting as a negative regulator of the mTOR pathway [51]. Importantly, the mTOR pathway is the final and common pathway that is also dysregulated in other polyposis syndromes caused by germline mutations in the PTEN, BMPR1A, and SMAD4 genes.
Inactivation of the STK11 gene is the cause of the development of hamartomas, but the role of hamartomatous polyps in the development of cancer has not been fully established. The possibility of malignancy with a tendency toward the sequence: hamartoma—dysplasia—cancer is discussed [52]. This assumption is supported by findings of adenoma foci within polyps, as well as cancers arising within a polyp [53]. Of interest are the data on the detection of somatic mutations of the STK11 gene in melanoma and non-small cell lung cancer in patients who are not carriers of the syndrome [54].
Mutations in the STK11 gene are detected in 70-80% of patients with Peutz-Jeghers syndrome. Differences in the diagnosis of mutations among patients may be due to the methods used to detect them. In addition, for patients with the syndrome phenotype, but without identified mutations in the STK11 gene, heterogeneity and the possibility of the existence of a second gene responsible for the syndrome at the 19q13.4 locus are discussed [55]. Genotype-phenotype correlation is important for monitoring patients with Peutz-Jeghers syndrome. Patients with missense mutations had a later onset of symptoms of the disease; nonsense mutations lead to a more severe course of the syndrome compared to other mutations in the STK11 gene [56]. Mutations in exon 6 of the STK11 gene carry a high risk of cancer for a patient [57]. Hotspots have been identified that are predominantly affected in patients with germline mutations in STK11, one in codon 51−84 of exon 1 and also in exon 7. However, to date, there are no clear differences between patients with STK11 mutations and those who do not have them. was not detected [58].
Clinical diagnosis of neoplasia in Peutz-Jeghers syndrome. As with FAP syndrome, the risk of its inheritance by offspring is high and amounts to 50%. Dynamic monitoring of patients who are carriers of the syndrome has two main goals: to prevent complications associated with polyps in the digestive tract (bleeding/anemia, intussusception, etc.) and to detect cancer at an early stage. Clinical diagnosis of cancer is comprehensive, focused on identifying polyps and neoplasia of the gastrointestinal tract, female reproductive organs, testicular neoplasia. Polyp dysplasia is diagnosed at an early age in 18% of patients. Colonoscopy is recommended starting at age 8 years, and if polyps are detected, endoscopy should be performed every 2 to 3 years along with observation of clinical symptoms. If no polyps are found, endoscopic diagnosis begins at age 18 at 2-5 year intervals [58]. Endoscopic examination of the stomach and esophagus, starting from the age of 10, every 2 years. The main tactic in endoscopic examination of the digestive tract is to remove all polyps larger than 1.5 cm in diameter [59].
Breast cancer screening uses an annual medical examination and self-examination. Ultrasound and mammography every 2-3 years, starting from the age of 20-25 years, after 40 years annually. Gynecological examination, including transvaginal ultrasound, CA125 analysis, annually from the age of 20. It should be taken into account that men can develop testicular Sertoli cell tumor (LCST), which secretes estrogen and leads to breast enlargement (gynecomastia) and abnormal physique. Therefore, regular examination of the testicles and ultrasound of the testicles in children and, if nothing is found, ultrasound of the abdominal organs and testicles annually starting from 20 years of age [59].
Diagnostics
Peutz-Jeghers syndrome is diagnosed using a biopsy. If a hamartomatous component is detected in a part of a polyp taken for analysis, then this is a typical symptom for this disease. Polyps ranging in size from 1 to 5 mm usually do not interfere with the normal functioning of the gastrointestinal tract. But as they grow, they can lead to bleeding, so growths larger than 1 cm must be removed. Polyps are characterized by moderate growth; they can be either multiple or single. With multiple cases, treatment is much more difficult. They cannot be removed once, so a gentle diet is used, as well as drug therapy aimed at slowing down the growth of tumors.
Other important diagnostic criteria are heredity and mucosal pigmentation. Since polyps are found in patients over 10 years of age, for children pigmentation on the mucous membranes is the main sign when making a diagnosis. Petz-Jeghers syndrome is also quite often accompanied by McCune-Albright syndrome (early sexual development). If a child has earlier pubertal development, the likelihood of him developing hamartomatous polyposis is quite high.
Stomach polyp - symptoms and treatment
Based on the number of growths there are:
- single polyps;
- multiple polyps;
- gastric polyposis (more than 20 polyps).
According to the clinical course, some Soviet scientists identified the following forms of gastric polyps:
- asymptomatic form;
- gastritis form;
- anemic form;
- complicated form (bleeding of the polyp and its prolapse into the duodenum);
- combined form (the appearance of a polyp and stomach cancer).
Based on endoscopic features, four types of gastric polyps can be distinguished:
- Type I - flat, raised, with indistinct edges;
- Type II - protruding, semicircular, with fairly clear boundaries;
- Type III - clearly protruding, rounded, with a retracted base;
- Type IV - pedunculated.
All of the above classifications deserve attention. However, in practice, the most important classification is based on the signs of degeneration of polyps into a malignant tumor [1][3].
According to the WHO classification, benign stomach tumors are defined as adenomas (adenomatous polyps). They are divided into papillary and tubular forms. Hyperplastic polyps are separately identified and included in the group of tumor-like processes [2][10].
In 2010, the British Society of Gastroenterology proposed its classification of gastric polyps, and also developed recommendations for patient management for each type of gastric polyp. According to this classification, gastric polyps are divided into five groups:
- Fundic gland polyps.
- Hyperplastic polyp.
- Adenomatous polyp.
- Hamartomatic polyps (associated with developmental defects):
- juvenile polyp;
- Peutz-Jeghers syndrome;
- Cowden's syndrome.
- Polyposis syndromes (nehamartoma):
- juvenile polyposis;
- familial adenomatous polyposis.
Fundic gland polyps are cystic expansions of the stomach's own glands and account for 16-51% of benign polyps. They usually reach 1-5 mm in diameter and are located mainly in the body or fundus of the stomach. They have a smooth, even surface, can be lobulated, and are covered with an unchanged mucous membrane. They can appear as an independent disease or as part of familial adenomatous polyposis of the colon.
Not associated with gastritis and Helicobacter pylori infection. They can form due to long-term use of proton pump inhibitors (drugs that reduce the production of hydrochloric acid). These medications increase the activity of gastrin (a stomach hormone), which stimulates the growth of epithelial cells.
The average development interval for fundic gland polyps is 32.5 months. Regression occurs three months after stopping taking proton pump inhibitors [4][7].
Hyperplastic polyps account for 30-93% of benign gastric polyps. It can be sessile and pedunculated, less than 2 cm in diameter. It is characterized by enlarged gastric pits, dilated and tortuous glands, and chronic inflammation of the gastric mucosa. A single polyp is most often located in the antrum (lower) part of the stomach. Multiple polyps can occur in all parts of the stomach.
Associated with chronic (Helicobacter-associated), chemical and atrophic gastritis. It occurs due to increased cell renewal (their layering on top of each other) in response to damage to the gastric epithelium (usually with erosions or gastric ulcers).
The hyperplastic polyp itself rarely becomes malignant (malignant), but it increases the risk of malignancy of the surrounding inflamed stomach tissue. Therefore, when a hyperplastic polyp is detected, it is recommended to perform a biopsy of the surrounding tissue (from 4-5 different places) [4][5].
Adenomatous polyp is a precancerous disease with a high potential for degeneration into cancer, especially when the polyp is more than 2 cm. It accounts for 3-26% of gastric polyps. Usually single, can be localized in any part of the stomach, but is more often found in the antrum (lower part of the stomach). The structure is tubular, villous and mixed. It occurs against the background of atrophic gastritis and intestinal metaplasia (when the gastric epithelium is replaced by intestinal epithelium) [4].
It is worth noting that in one study at the Vitebsk Regional Clinical Hospital, a hyperplastic polyp with areas of adenomatosis was found. The authors suggested that hyperplastic polyps can transform into adenomatous ones. They also allowed themselves to identify another histological form of polyps - a hyperplastic polyp with focal adenomatosis. It is a true benign tumor of the stomach, capable of developing into cancer [10].
Hamartomatic polyps are rare, but it’s worth saying a few words about them.
A single juvenile (youthful) polyp has no malignant potential, but, like all polyps, it can be complicated by bleeding or strangulation, since such polyps are mainly located in the lower part of the stomach and are susceptible to trauma.
Peutz-Jeghers syndrome is a rare hereditary disease that is accompanied by the appearance of hamartomatous polyps in the gastrointestinal tract, as well as pigmentation in the lips, fingers and buccal mucosa. With this disease, there is a high risk of malignancy of both the digestive organs and the lungs, mammary glands, pancreas, and uterus. Therefore, such patients should be closely monitored.
Cowden's syndrome is a rare hereditary disease that is accompanied by the presence of polyps of the gastrointestinal tract, benign tumors of the mouth, as well as malformations of various organs (breast, thyroid and genitals). Polyps with this syndrome very rarely degenerate into cancer, but still require observation.
Polyposis syndromes include juvenile polyposis and familial polyposis syndrome.
In juvenile polyposis, many juvenile polyps are found that have malignant potential. This disease can also be complicated by gastrointestinal bleeding and enteropathy, a disease of the small intestine that is accompanied by loss of protein and other nutrients.
Familial polyposis syndrome is a hereditary disease with a very high risk of cancer of the stomach and other parts of the gastrointestinal tract. Affected family members have a large number of colonic and rectal adenomas, which have a 100% chance of developing into cancer unless a prophylactic colectomy—complete removal of the colon—is performed. Gastric polyps are detected in 30-100% of cases. This syndrome does not have a clear connection with gastritis caused by Helicobacter [4].
Specific stages of disease development are not distinguished, since different types of polyps have different origins and development. But if we talk about frequently occurring adenomatous and hyperplastic polyps, then we can conditionally distinguish three stages of development [1]:
- polypous gastritis (inflammation or atrophy of the gastric mucosa);
- stomach polyp;
- stomach cancer.
Pates-Jeghers syndrome
"Peutz-Jeghers syndrome"
— genetic study to identify pathogenic mutations in the STK11 gene associated with the risk of developing Peutz-Jeghers syndrome. The panel was developed based on the American Cancer Association clinical guidelines version 1.2019. Peutz-Jeghers syndrome is a disease with an autosomal dominant type of inheritance (that is, for its development a mutation in one of the parents is sufficient), characterized by the development of benign neoplasms called hamartomatous polyps in the gastrointestinal tract (especially in the stomach and intestines) and significantly increased risk of developing certain types of cancer, as well as characteristic pigmented areas on the skin and mucous membranes. In most patients, they appear during the first year of life and often disappear as the person gets older. Polyps can cause health problems such as occasional bowel obstruction, chronic bleeding, and abdominal pain. Most cases of this disease are caused by a mutation in the STK11 (LKB1) gene.
The STK11 protein (serine-threonine kinase 11) reduces the rate of too rapid cell growth and division and is involved in the initiation of apoptosis (activates programmed cell death). Mutations in the gene are associated with uncontrolled cell growth and division, which leads to the formation of benign polyps and cancerous tumors.
The study includes the identification of 14 polymorphisms of the STK11 gene
1. STK11 Serine-threonine kinase 11 (g.1221237C>A; c.759C>A; p.Tyr253Ter; rs137853075) 2. STK11 Serine-threonine kinase 11 (g.1221321del; c.843del; p.Leu282SerfsTer5; rs587 776656) 3. STK11 Serine-threonine kinase 11 (g.1220701_1220704del; c.718_721del; p.Ser240LeufsTer46; rs587776657) 4. STK11 Serine-threonine kinase 11 (g.1220372G>A; c.465-1G>A; rs587776658) 5. STK11 Serine-threonine kinase 11 (g.1207163A>T/G; c.250A>T/G; p.Lys84Ter/Glu; rs137853076) 6. STK11 Serine-threonine kinase 11 (g.1221312_1221313del; c.834_835del; p. Cys278TrpfsTer6; rs587776659) 7. STK11 Serine-threonine kinase 11 (g.1207113T>C; c.200T>C; p.Leu67Pro; rs137853077) 8. STK11 Serine-threonine kinase 11 (g.1221994_12220 02del;c.908_916del;p. Ile303_His306delinsAsn; rs587776660) 9. STK11 Serine-threonine kinase 11 (g.1207082G>T; c.169G>T; p.Glu57Ter; rs137854584) 10. STK11 Serine-threonine kinase 11 (g.1219367del ; c.418del; p. Leu140TrpfsTer21;rs397518440) 11. STK11 Serine-threonine kinase 11 (g.1207110dup; c.197dup; p.Leu67AlafsTer96; rs397518441) 12. STK11 Serine-threonine kinase 11 (g.1220700G>C; c.717G>C; p.Trp239Cys; rs137853082) 13. STK11 Serine-threonine kinase 11 (g.1221977del; c.891del; p.Arg2 97SerfsTer39; rs587776661) 14. STK11 Serine-threonine kinase 11 (g.1221216C>G; c.738C>G; p.Tyr246Ter; rs137853083)
Indications
Determination of genetic predisposition to the development of malignant tumors and diagnosis of Peutz-Jeghers syndrome. And also for early detection of mutations in relatives of patients with this syndrome.
Preparation
Taking venous blood samples for DNA testing can be done at any time of the day on an empty stomach (8-12 hours, but no more than 14 hours) or no earlier than 4 hours after a light meal; a special diet and medication will not affect the result. It is acceptable to drink clean water without gas and sugar. Avoid food overload the day before. Avoid drinking alcohol at least 24 hours before taking blood.
Please note that the results of the DNA test may be affected by a recent blood transfusion or bone marrow transplant (in this case, DNA testing is not recommended for at least 1 month after the transplant or blood transfusion).
Interpretation of results
To interpret the results of genetic testing, consultation with a geneticist is required.