Одним из наиболее тяжелых недугов пищеварительной системы, который имеет генетический характер, является диффузный полипоз, поражающий прямую и толстую кишку. Эту болезнь следует отличать от поражения одиночными и групповыми полипами, поскольку, поражая человека еще в молодости, к 50 годам она практически в 100 процентах случаев переходит в онкологию. Заболевание может протекать в трех основных формах:
- Как диффузный семейный полипоз;
- Синдром Гарднера;
- Синдром Пейтца-Егерса.
Именно о последней разновидности мы и поговорим более подробно, в частности, узнаем, что становится причиной ее развития, рассмотрим симптомы болезни и методы ее лечения.
Всему причиной наследственность
Говоря о причинах развития указанного синдрома, специалисты выделяют исключительно наследственную предрасположенность. Статистика неутешительна: если в семье человека есть родственники, которые страдают недугом, он заболеет почти со стопроцентной вероятностью. Точнее, болезнь уже будет заложена в нем с рождения, и со временем, как правило, в достаточно юном возрасте, она проявится.
Казалось бы, все достаточно понятно. На самом же деле медики считают этот недуг одним из наиболее малоизученных. Все дело в том, что пока исследователям удалось выявить лишь один ген, наличие которого и определяет человека в группу риска. Но медики предполагают наличие и ряда других генов, выявить которые пока не удается.
Синдром Пейтца-Егерса (Гамартомный полипоз)
Постановка диагноза зачастую затруднена, что обусловлено низкой распространенностью синдрома Пейтца-Егерса и неспецифичностью диспепсических симптомов. Заподозрить заболевание можно при обнаружении у пациента выраженной пигментации, которая появляется уже в раннем возрасте, наличии данных о наследственной отягощенности. Диагностический поиск предполагает проведение комплексного обследования пищеварительной системы. Наиболее информативными являются:
- Сонография брюшной полости
. Неинвазивное ультразвуковое сканирование применяется для экспресс-диагностики синдрома. УЗИ органов брюшной полости позволяет обнаружить объемные образования в просвете кишечника и патологию других органов ЖКТ. Сонография может быть недостаточно информативной при небольших размерах полипов (до 5 мм). - Контрастная рентгенография.
Выполнение рентгенограмм после перорального введения густой бариевой взвеси необходимо для выявления полиповидных разрастаний. При аномалии Пейтца-Егерса просматриваются дефекты наполнения округлой формы. Прогностически неблагоприятно обнаружение нечетких контуров или локального нарушения перистальтики. - Эндоскопическое исследование
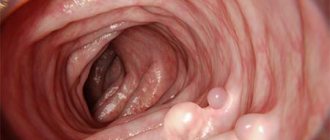
. Для визуализации слизистой оболочки верхних отделов пищеварительной трубки осуществляют ЭГДС, для изучения состояния толстой кишки проводят колоноскопию. Полипы Пейтца-Егерса по внешнему виду напоминают аденомы, могут достигать нескольких сантиметров и иметь ножку. Иногда отмечают гиперемию и эрозии эпителия. - Гистологический анализ
. Цитоморфологическое изучение биоптатов необходимо для дифференцировки с другими новообразованиями. Для заболевания характерно сохранение всех слоев слизистой оболочки, отсутствие клеточной атипии и патологических митозов, разрастание стромы и гладкомышечных волокон. По тканевой структуре полипы являются гамартомами.
В общем анализе крови обнаруживают признаки анемического синдрома – уменьшение количества гемоглобина и числа эритроцитов. В биохимическом анализе крови выявляют снижение уровня общего белка. Всем пациентам выполняют копрограмму, для подтверждения кровотечений из ЖКТ проводят реакцию Грегерсена на скрытую кровь. По показаниям производят цитологический анализ кожных биоптатов из пигментированных участков.
Дифференциальный диагноз при симптомокомплексе Пейтца-Егерса в первую очередь осуществляют с другими формами полипоза (диффузным семейным, ювенильным). Основными диагностическими отличиями являются наличие гиперпигментации отдельных участков кожного покрова и специфическое гистологическое строение полипов. Также нужно дифференцировать заболевание с множественным лентиго. Для обследования пациента с признаками полипозного синдрома привлекаются специалист-гастроэнтеролог, проктолог, дерматолог.
Стандартные методы лечения
В первую очередь больным следует получить консультацию клинического генетика или генетического консультанта. Поскольку от синдрома Пейтца-Егерса нет лекарства, лечение в основном направлено на наблюдение и контроль над симптомами. После первоначального диагноза людям старше 8 лет или имеющим симптомы рекомендуется пройти эндоскопическое обследование и обследование тонкой кишки. Последнее может быть выполнено с помощью магнитно-резонансной томографии кишечника (магнитно-резонансная энтерография, МР-энтерография) или путем проглатывания капсулы, которая делает внутренние изображения из желудочно-кишечного тракта (видеокапсульная эндоскопия). Гинекологическое обследование и обследование груди также рекомендуется женщинам старше 18 лет. Мужчинам рекомендуется обследование яичек.
После первоначального обследования после постановки диагноза каждые 2–3 года следует проводить эндоскопию, колоноскопию и обследование тонкой кишки для выявления полипов и потенциальных опухолей. Женщинам рекомендуется делать ежегодно маммографию. Мужчинам УЗИ яичек можно делать раз в два года.
Поскольку синдром Пейтца-Егерса увеличивает риск рака груди, матки и яичников, больные женщины могут пройти профилактическую мастэктомию, гистерэктомию или сальпингоофорэктомию (хирургическое удаление груди, матки, маточных труб и яичников соответственно).
Полипы размером более 1 см удаляются эндоскопическими методами, чтобы избежать связанных с полипами осложнений, таких как кровотечение и инвагинация. Эти осложнения могут потребовать хирургического вмешательства. Если пациенту предстоит операция, эндоскопическое удаление полипов (полипэктомия) выполняется одновременно с операцией, чтобы снизить риск рецидива осложнений и хирургического вмешательства.
В случаях, когда темные пигментные пятна (меланоцитарные пятна) оказывают крайне негативное психологическое воздействие на пораженных людей, их можно частично удалить с помощью лазерной терапии.
Лечение
Несмотря на то что еще в 1921 году Пейтц описал синдром Пейтца — Егерса — Турена, симптомы и лечение этого заболевания еще недостаточно изучены. На данный момент комплексное лечение данного заболевания не разработано. Поскольку при синдроме в кишечнике вырастают сотни полипов, профилактически удалить их все невозможно из-за высокого риска такой операции. Потому больные синдромом Пейтца — Егерса в течение жизни переносят множество хирургических вмешательств по удалению полипов, когда те достигают критических размеров.
Дерматозы параонкологические
ДЕРМАТОЗЫ ПАРАОНКОЛОГИЧЕСКИЕ
Дерматозы параонкологические (син.: дерматозы паранеопластические) — кожные реакции на злокачественные новообразования внутренних органов и систем, которые изменяют физиологические процессы в организме и приводят к накоплению в нем обычно биологически активных веществ (гормоны, факторы роста и др.) и опухолевых антигенов, способствующих возникновению новых клинических симптомов.
Поражения кожи и слизистых при этом можно разделить на три группы:
- наследственные синдромы, при которых злокачественная опухоль сочетается с заболеванием кожи и слизистых;
- паранеопластшеские дерматозы, обусловленные влиянием опухоли на метаболизм, иммунитет и регуляторные системы организма;
- метастазирование злокачественных опухолей в кожу и слизистые.
Наследственные синдромы Гарднера синдром
Гарднера синдром — наследственный симптомокомплекс, включающий в себя эпидермоидные кисты, фибромы, остеомы в сочетании с полипозом прямой кишки.
Тип наследования: аутосомно-доминантный.
Преобладающий возраст. Развивается в возрасте 4—10 лет, редко позднее.
Клиническая картина
- Эпидермоидные кисты на коже лица, волосистой части головы, конечностях, реже груди.
- Остеомы развиваются главным образом в челюстных и клиновидных костях.
- Полипоз ободочной и прямой кишок, но нередко полипы обнаруживают в тонкой кишке и желудке. Полипы в толстом кишечнике имеют тенденцию к малигнизации.
Диагностика основывается на клинических данных и специальных методах обследования пищеварительного тракта.
Лечение. Хирургическое удаление полипов.
Ассоциирующееся злокачественное новообразование: рак желудочно-кишечного тракта, а также костей и эндокринных желез.
Каудена синдром
Каудена синдром — редкий наследственный симптомокомплекс. Характеризуется множественными гамартомами экто- и мезодер-мального происхождения в сочетании с предрасположенностью к злокачественным опухолям, особенно молочных и щитовидной желез. Синдром назван фамилией семьи, в которой впервые были отмечены такие больные.
Преобладающий возраст. 4—75 лет (у половины больных до 40 лет); мужчины болеют чаще.
Тип наследования: аутосомно-доминантный с вариабельной экспрессивностью гена.
Клиническая картина. Постоянным симптомом заболевания, варьирующим лишь в своей интенсивности, являются слизисто-кож-ные бородавчато-папилломатозные поражения. Вначале возникают множественные, цвета кожи лихеноидные папулы (до 4 мм в диаметре), на веках, вокруг глаз и ротового отверстия. В дальнейшем они сливаются и образуют в указанных зонах поверхности, напоминающие булыжную мостовую. Процесс может захватить губы, слизистую оболочку щек, глотки, гортани. На ладонях, подошвах с переходом на пальцы — небольшие полупрозрачные кератотические наслоения. Возможны также ангиомы, липомы, меланома.
Общее состояние. Изменения кожи обычно сочетаются с фиброзным кистозом или фиброаденомой молочных желез у женщин с наклонностью к озлокачествлению (30% случаев) и зобом, аденомой или раком щитовидной железы. Реже встречаются другие аномалии (готическое небо, дисхромия кожи, нарушения скелета, полипоз пищеварительного тракта, гинекологическая патология, первичный Т-клеточный иммунодефицит).
Диагностика основывается на клинических и гистологических данных.
Дифференциальный диагноз
- с болезнью Дарье;
- туберозным склерозом.
Лечение
Хирургическое удаление новообразований, диспансерное наблюдение с периодической маммографией и контролем состояния щитовидной железы. Избегать применения эстрогенов.
Течение и прогноз. Новые опухоли могут возникать в течение всей жизни.
Синоним: синдром множественной гамартомы и неоплазии.
Ассоциирующееся злокачественное новообразование: аденокарци-нома молочной железы.
Нейрофиброматоз (см. с. 115)
Ассоциирующееся злокачественное новообразование:
- нейрофибросаркома;
- феохромоцитома;
- рабдомиосаркома и др.
Синдром Пейтца—Турена-Егерса
Основной признак этого наследственного синдрома — мелкие множественные пигментные пятна (лентиго) на губах и слизистых рта. Полипоз желудка, тонкого и толстого кишечника. Полипы представляют собой гамартомы, встречаются у большинства больных.
Тип наследования: аутосомно-доминантный.
Пол
•Лентиго обнаруживают у грудных детей и в младшем детском возрасте./>
•Полипы — у детей старшего возраста и взрослых до 30 лет. Мужчины и женщины болеют одинаково часто./>
Больные предрасположены к раку молочной железы, яичников и поджелудочной железы.
Течение. Лентиго встречается уже при рождении или появляется в грудном и младшем детском возрасте.
Клиническая картина. На коже губ, слизистых щек, лица, вокруг естественных отверстий, ладоней, подошв появляются пятна, округлой и овальной формы, размером 2—5 мм темно-коричневого или черного цвета. Высыпания на лице мельче, чем на ладонях, подошвах и слизистых. Пятна на губах со временем могут исчезнуть, но пигментация слизистой сохраняется на всю жизнь. Имеются полипы тонкой, реже — толстой кишки. Возможны инвагинация и кишечная непроходимость.
Диагностика
- Анамнез.
- Клиническая картина.
- Выявление полипов ЖКТ.
Лечение не разработано. Проводят хирургическое удаление кровоточащих полипов или превышающих в размерах 1,5 см. Синонимы
- Периорифициальный лентигиоз.
- Синдром Пейтца-Егерса.
• Полипоз пигментно-пятнистый. Ассоциирующееся злокачественное новообразование:/>
- рак молочной железы;
- рак яичников;
- рак поджелудочной железы.
Паранеопластические дерматозы облигатные (всегда сочетаются со злокачественной
опухолью)
Acanthosis nigricans
Ассоциирующееся злокачественное новообразование:
- аденокарцинома желудка;
- рак матки;
- яичников;
- предстательной железы;
•легких;
• лимфоретикулярной системы. Акрокератоз псориазиформный Базекса/>
Акрокератоз псориазиформный Базекса — параонкологический дерматоз.
Пол. Чаще встречается у мужчин.
Клиническая картина. Характеризуется эритематосквамозными очагами поражения с акральной локализацией. Кожные изменения
с шелушением на коже носа, ушных раковин, кистей, стоп. Постепенно сыпь становится более генерализованной. Возникают дистрофия ногтей, паронихии. Изменения кожи лица могут быть похожи на проявления экземы или красной волчанки, в то время как акральные участки поражения (на кистях, стопах) больше напоминают псориаз.
Гистологически выявляют гиперкератоз, паракератоз, фокальный спонгиоз и воспалительный смешанный инфильтрат в дерме.
Диагностика проводится на основании клинических и гистологических данных.
Дифференциальный диагноз проводят с себорейной экземой, аллергическим дерматитом, синдромом Райтера, псориазом, красной волчанкой.
Лечение: выявление и радикальное удаление злокачественного новообразования, этретинат внутрь (0,5 мг/кг в сутки).
Ассоциирующееся злокачественное новообразование:
• рак верхних дыхательных путей;
- пищеварительного тракта.
Карциноидный синдром
Внезапная временная эритема темно-красного или пурпурного цвета в результате притока крови к лицу и шее; диарея, спазмирующая боль в животе, правожелудочковая недостаточность, астма, отеки, телеангиэктазии, цианоз, пеллагроподобные высыпания, признаки синдрома Кушинга. В моче — увеличение уровня метаболита серотонина 5-гидрокси-индолацетиновой кислоты.
Ассоциирующееся злокачественное новообразование: карциноидная опухоль в пищеварительном тракте, чаще в тонкой кишке, легком, метастазы в печени
Эритема гирляндообразная мигрирующая Гаммела
Эритема гирляндообразная мигрирующая Гаммела — облигатный параонкологический дерматоз, развитие которого связывают с аутоиммунным механизмом воздействия тканей опухоли. Описана Gammelв 1952 г. Наблюдается чаще за несколько месяцев (иногда за 2—4 года) до клинических признаков злокачественного новообразования внутренних органов.
Клиническая картина. Поражение кожи характеризуется множественной пятнистой, иногда несколько отечной, сыпью, первые элементы которой появляются обычно на лице, а затем распространяются на кожу туловища, проксимальных отделов конечностей в виде причудливых фигур, колец, гирлянд, быстро перемещающихся по коже (в течение нескольких часов или дней). Возможны мелкопластинчатое шелушение, легкий зуд. Ладони и подошвы не поражаются. Клинически и гистологически заболевание напоминает кольцевидную эритему Дарье.
Диагностика основывается на клинических данных. Необходимо тщательное онкологическое обследование пациента.
Лечение: радикальное удаление злокачественного новообразования внутренних органов приводит к полному регрессу поражения кожи.
Ассоциирующееся злокачественное новообразование:
- рак легких;
- молочных желез;
- половых органов;
- желудка.
Некролитическая эритема
Некролитическая мигрирующая эритема обусловлена развитием глюкагономы — опухолью, растущей из ое-клеток островков поджелудочной железы. Опухоль продуцирует глюкагон и сопровождается весьма характерными высыпаниями.
Этиология. Большинство случаев некролитической мигрирующей эритемы обусловлены избытком глюкагона в крови, однако патогенез ее неизвестен.
Возраст. Средний и пожилой.
Клиническая картина. Воспалительные бляшки с периферическим ростом и эпителизацией в центре. Бляшки сливаются, располагаясь в виде колец, дуг, что придает очагам поражения вид географической карты, по краям которых образуются пузырьки, корки и чешуйки. Располагаются высыпания на сгибательной поверхности конечностей, в крупных складках, вокруг естественных отверстий. Кончики пальцев рук красные, блестящие, с эрозиями.
Методы исследования. Повышение уровня глюкагона плазмы натощак более 1000 нг/л.
Диагностика
- Клиническая картина.
- Результаты биопсии.
- Определение уровня глюкагона в крови.
Дифференциальный диагноз
- Энтеропатический акродерматит.
- Экссудативный псориаз.
- Кандидоз.
- Доброкачественная семейная пузырчатка Гужеро—Хейли—Хейли.
Лечение. Хирургическое удаление опухоли.
Ассоциирующееся злокачественное новообразование:
- глюкагонома;
- тяжелое течение цирроза печени;
- рак легкого.
Педжета болезнь молочной железы
Педжета болезнь молочной железы — внутрипротоковый рак грудной железы. Может ограничиваться поражением эпидермиса молочной железы или сочетаться с карциномой молочной железы, при этом эпидермис поражается вторично.
Возраст, пол. Встречается главным образом у женщин 50—60 лет.
Клиническая картина. Заболевание характеризуется очагом застойной гиперемии с четкими границами в области ореолы и соска молочной железы. На поверхности очага могут наблюдаться признаки экзематизации: шелушение, эрозии, мокнутие, серозные и геморрагические корки, зуд. Возможны вьщеления из соска серозного или геморрагического характера. Постепенно очаг уплотняется, захватывает кожу грудной клетки, его границы приподнимаются. Сосок втягивается или исчезает, а в его глубине определяется участок уплотнения; могут увеличиваться регионарные подмышечные лимфатические узлы. Течение неуклонно прогрессирующее. Первичный рак молочной железы распространяется книзу до эпителия долек, а также кверху и кнаружи до эпидермиса, где развивается поражение кожи. В поздней стадии болезни раковые клетки проникают через стенку протока или дольки и инфильтрируют соединительную ткань молочной железы — развиваются лимфогенные метастазы.
Диагноз ставят на основании клинической картины, течения процесса, результатов гистологического исследования.
Дифференциальный диагноз с
- экземой сосков;
- болезнью Боуэна;
- поверхностной формой базалиомы;
- меланомой.
Лечение: хирургическое удаление опухоли.
Ассоциирующееся злокачественное новообразование: аденокарцинома молочной железы внутрипротоковая.
Факультативные (часто сочетаются со злокачественной опухолью)
Педжета болезнь экстрамаммарная
Педжета болезнь экстрамаммарная — злокачественная опухоль, поражающая перианальную область, наружные половые органы и подмышечные впадины.
Возраст, пол. Встречается как у женщин (чаще), так и у мужчин среднего и пожилого возраста.
Клиническая картина характеризуется красными пятнами или бляшками с неровными очертаниями, мацерированной поверхностью, корковыми наслоениями, зудом. Часто имеет тенденцию к ин-вазивному росту и трансформации в плоско клеточный рак. Нередко ассоциируется со злокачественными новообразованиями внутренних органов. Гистологически не отличается от маммарной локализации болезни Педжета, но протяженность патологического процесса значительно превышает видимые размеры очага поражения, что обусловливает частые рецидивы после лечения.
Диагноз основывается на клинических и гистологических данных.
Дифференциальный диагноз с
•болезнью Боуэна;
- педжетоидной эпителиомой;
- экземой.
Тактика ведения. Обязательно исследуют прямую кишку, шейку матки и мочеиспускательный канал в поисках первичной опухоли.
Лечение
- Хирургическое иссечение с широким захватом внешне не измененной кожи.
- Назначают блеомицин по 15 мг в/м ежедневно; на курс 5—6 мг/кг, интервалы между курсами 1,5—2 мес, или проспи-дин в/м или в/в по 0,2 г/сут.; на курс 3—4 г, а в тяжелых случаях 5—6 г.
- При поверхностных формах, локализующихся на половых органах или в промежности, проводится наружная химиотерапия: 5% фторурациловая, фторофуровая, 30—50% проспидино-вая, 0,5% колхаминовая мази.
- Используются также криодеструкция и лазеротерапия.
Ассоциирующееся злокачественное новообразование:
- рак матки;
- рак яичников;
- рак предстательной железы;
- рак прямой кишки.
Боуэна болезнь
Боуэна болезнь — внутриэпидермальный кожный рак, обязательно трансформирующийся в плоскоклеточный.
Возраст, пол. Чаще развивается у пожилых людей (70—80 лет) обоего пола.
Этиология и патогенез. Развитие заболевания обусловлено действием УФ-облучения, длительной травматизацией кожи, при локализации процесса на закрытых участках кожи — контактом с солями мышьяка (лекарственные препараты, промышленное производство и др.). Очаг чаще располагается на туловище, верхних конечностях, в области промежности, иногда на слизистой оболочке рта.
Клиническая картина. Процесс представлен солитарной плотной бляшкой неправильной или округлой формы, покрытой белыми или желтоватыми чешуйками, которые легко удаляют с образованием эрозий и мокнутием, но без признаков кровотечения. Эрозии могут быть покрыты серозными и геморрагическими корками. Размер очага — от 2 мм до ладони ребенка. К важным особенностям болезни Боуэна относятся неравномерный рост очага по периферии, его пестрота за счет участков атрофии, эрозирования, гиперкератоза, бородавчатых разрастаний, возвышение краевой зоны.
Течение и прогноз. Течение медленное, неуклонно прогрессирующее. Плоскоклеточный рак может развиться даже на ранних стадиях болезни. Рецидивы возникают как результат неадекватного лечения.
Диагноз устанавливают на основании клинической картины и результатов гистологического исследования.
Дифференциальный диагноз проводят с экземой, псориазом, бородавчатым туберкулезом кожи, плоскоклеточным и метатипичес-ким раком, солнечным кератозом, старческой кератомой, базалиомой, бовеноидным папулезом.
Лечение: при небольших размерах очага (до 2 см) используют аппликации 30—50% проспидиновой, 5% фторурациловой мазей, при диаметре очага более 2 см показаны криодеструкция, хирургическое и электрохирургическое лечение; удаление углекислым лазером. Если очаг локализуется на слизистой оболочке, назначают 5% фторурациловую мазь, внутрь — ароматические ретиноиды (тигазон из расчета 1 мг/кг в сутки в течение 1—2 мес). Рентгенотерапия малоэффективна.
Ассоциирующееся злокачественное новообразование:
- аденокарцинома желудка;
- рак матки;
- яичников;
- предстательной железы;
- легких;
- лимфоретикулярной системы. Д
Дерматит эксфолиативный
Дерматит эксфолиативный подострый генерализованный — специфическая эритродермия, являющаяся проявлением Т-клеточной лимфомы кожи. Развитию заболевания способствуют нарушения иммунитета.
Клиническая картина. Чаще всего эритродермии предшествует появление очагов поражения, напоминающих экзему или нейродермит. Сформировавшаяся клиническая картина характеризуется генерализованной эритемой, охватывающей весь кожный покров, отечностью кожи, обильным крупнопластинчатым шелушением, а также ладонно-подошвенным кератозом, выпадением волос и ногтей, зудом, ознобами, увеличением периферических лимфатических узлов. В крови — лейкоцитоз, моноцитоз, эозинофилия и повышенная СОЭ.
Синоним: эритродермия Вильсона—Брока.
См. Лимфомы кожи.
Ассоциирующееся злокачественное новообразование: лимфопроли-феративные заболевания.
Зуд кожный
Ассоциирующееся злокачественное новообразование:
- лимфома;
- лейкоз.
Вероятные (иногда сочетаются со злокачественной опухолью) Ихтиоз приобретенный Ассоциирующееся злокачественное новообразование:
- лимфогранулематоз;
- саркома яичек.
Пемфигоид буллезный
Ассоциирующееся злокачественное новообразование: злокачественные опухоли различных типов и локализации. .
Герпетиформный дерматит Дюринга
Ассоциирующееся злокачественное новообразование:
- аденокарцинома желудка;
- рак молочной железы и др.
Эритема кольцевидная центробежная Дарье
Ассоциирующееся злокачественное новообразование:
- аденокарцинома желудка;
- рак молочной железы;
- рак легкого.
Метастазы в кожу
Метастазы злокачественных опухолей представляют собой одиночные или множественные узлы, локализующиеся в коже или подкожной клетчатке. По механизму возникновения различают гематогенные, лимфогенные и имплантационные метастазы.
Частота. Метастазы в кожу находят у 0,7—9% всех больных злокачественными опухолями.
Преобладающий возраст. Любой, но чаще пожилой.
Течение и прогноз. Прогноз обычно неблагоприятный. Средняя продолжительность жизни больных после выявления метастазов в кожу всего несколько месяцев.
Элементы сыпи. Узел, фиброзная бляшка, плотной консистенции, выступающие над поверхностью кожи. Метастазы обычно обнаруживают, когда их размеры превышают 5 мм. Цвет высыпаний розовый или красный. Метастазы меланомы выглядят как внутри-кожные узлы синего, серого или черного цвета.
Виды поражения кожи
Рак молочной железы. При лимфогенном распространении опухоли на коже пораженной железы образуются воспаленные гиперемированные бляшки, как при роже, плотные уплощенные папулы и бляшки, телеангиэктазии или узлы.
Рак толстой кишки. Метастазирует в кожу живота, промежности, в кожу волосистой части головы и лица. Метастазы представлены воспалительными бляшками, реже узлами на ножке или широком оснований на ягодицах; группами обильно васкуляризированных образований в паховой области и на мошонке.
Рак легкого. Чаще дает метастазы в виде красноватых узлов, расположенных симметрично на волосистой части головы или туловище. Локализация высыпаний может совпадать с ходом межреберных сосудов.
Рак почки. Метастазы могут быть одиночными или множественными, обычно в виде сосудистых образований, похожих на телеангиэктатическую гранулему, иногда они располагаются на ножке.
Преимущественная локализация на голове и шее, реже — на туловище и конечностях.
Меланома. Метастазы меланомы представлены одиночными или множественными узлами темного цвета, но иногда встречаются и беспигментные формы. Метастазы и рецидивы меланомы могут появляться как в отдаленных участках кожи, так и в расположенных рядом.
Рак мочевого пузыря и рак яичников. Эти опухоли могут распространяться на кожу живота и паховой области, образуя воспалительные гиперемированные бляшки, напоминающие рожу.
Диагностика
- Наличие в анамнезе злокачественной опухоли внутренних органов.
- Гистологическое исследование.
Лечение. Если позволяет состояние больного, иссечение одиночных метастазов.
Риск онкологии
У пациентов с синдромом Пейтца — Егерса значительно выше риск развития рака ЖКТ, чем у здоровых людей. Чаще всего онкология поражает толстый кишечник, тонкий кишечник и поджелудочную железу. У женщин часто наблюдается онкология молочной железы (45 % случаев). Именно в высокой вероятности развития рака и заключается опасность заболевания, Пейтца — Егерса синдром нужно контролировать с помощью нескольких клинических процедур.
Учитывая повышенный риск развития онкологии, всем больным стоит проходить рентгенографию тонкого кишечника примерно раз в 2 года.
Гастроскопия и колоноскопия помогут лечащему врачу контролировать состояние желудка и кишечника, а также следить за размерами полипов. Даже при успешной операции по удалению полипа вероятность рецидива высока, потому эти обследования абсолютно необходимы.
Женщинам от 25 лет нужно проходить маммографию каждый год, чтобы заметить онкологию на ранней стадии.
В диагностике рака важную роль играет исследование на наличие в крови карциноэмбрионального антигена. Это химическое вещество, вырабатываемое при раке кишечника, онкологии молочной железы или легких.
Также важно определение и других антигенов рака (они же опухолевые антигены) – веществ, вырабатываемых в пораженных онкологией клетках любого органа. Характер их зависит от локализации опухоли, именно поэтому эти вещества играют важную роль в диагностике.
Большинство больных синдромом Пейтца — Эгерса не доживают до 60 лет, умирая от рака поджелудочной железы (11 %), желудка (57 %), кишечника (85 %), молочной железы (45 %). Несколько повышается и риск развития онкологии легких, яичек, шейки матки, яичников. Если онкология обнаружена на поздней стадии, она приводит к летальному исходу. Именно поэтому так важна своевременная диагностика.
Результаты
В некоторых генетически обусловленных синдромах злокачественные опухоли являются интегральной частью, примером является САП (ОMIM#175100). Этот синдром имеет аутосомно-доминантный тип наследования с распространенностью 1 случай на 10 000 новорожденных. Характеризуется появлением множества полипов, малигнизация которых наступает в 100% случаев и именно при этом синдроме возможна идентификация пораженных индивидов до появления рака. Клиническая картина заболевания включает три фенотипа, среди которых классическая форма синдрома имеет тяжелую и умеренно тяжелую формы. Тяжелая (классическая) форма САП: более 2000, 5000 полипов (или профузный полипоз) в левой половине кишечника, раннее появление полипов, быстрая малигнизация. Умеренная (классическая) форма: наличие сотен и более аденоматозных полипов, типично локализующихся также в дистальных отделах толстой кишки. Третья, слабовыраженная форма САП, называемая также «аттенуированной» или синдромом ACAП: небольшое число (более 20, но менее 100) аденом в большинстве случаев в правой половине толстой кишки и появление в более позднем возрасте (старше 15 лет). У 10% носителей первых двух форм синдрома полипы появляются в возрасте до 10 лет, а к 20 годам полипы развиваются у 95% пациентов, у большинства из них семейный анамнез отягощен. Аденоматозные полипы доброкачественные, но некоторые из них, если их хирургически не удалить, в среднем к 35 годам жизни трансформируются в колоректальный рак [1].
По нашим данным, наиболее ранняя диагностика малигнизации была у одного из пациентов с семейной формой САП, у которого полипы диагностированы в возрасте 4 лет, колонэктомия произведена в 14 лет. Рак оставшейся части прямой кишки, выявленный в процессе наблюдения, хирургически удален на ранней стадии в 21 год. На рис. 1 картина классической формы синдрома САП, на рис. 2 (а, б) представлена гистологическая и цитологическая картина аденомы при синдроме САП.
Рис. 1. Диффузный полипоз толстой кишки.
Рис. 2. Тубуловиллезная аденома при синдроме САП (а), гистологическая окраска гематоксилином и эозином, ×100; цитологический препарат (б).
Синдром САП — мультиопухолевый синдром, при котором пациенты имеют предрасположенность к появлению доброкачественных и злокачественных образований мезенхимального, эктодермального и эндодермального происхождения в разных органах. Так, например, врожденная гипертрофия пигментного эпителия сетчатки (ВГПЭС) глаза, эпидермоидные кисты, аномалии костей и зубов, остеомы не ассоциируют с малигнизацией. Злокачественный потенциал несут в себе десмоидные, адренокортикальные опухоли, полипы двенадцатиперстной кишки и желудка, аденомы печени. Опухоли щитовидной железы, яичников, панкреатобилиарной зоны, мозга и печени при этом синдроме часто манифестируют как злокачественные [2—6].
При третьей, слабовыраженной форме САП, внекишечные проявления встречаются редко, а риск развития рака толстой кишки зависит от тяжести поражения полипами. Полипы как наиболее постоянный компонент синдрома у пациентов с САП могут возникать в желудке, двенадцатиперстной кишке. Такие образования, как кожные фибромы и эпидермоидные кисты, липомы, множественные остеомы костей лица, чаще нижней челюсти, аномалии роста зубов, ювенильные назофарингеальные ангиофибромы, не нуждаются в активном лечении. Однако десмоидные опухоли могут быть клинически агрессивными, проявляться в виде одного или нескольких образований в брюшной полости или вдоль позвоночника, конечностей. Их совместное проявление с полипами желудочно-кишечного тракта, остеомами лицевых костей, кожными фибромами относят к синдрому Гарднера (вариант САП), при котором повышен риск развития рака, в том числе желудка.
У наших пациентов с синдромом Гарднера остеомы костей лица, кишечные полипы и полипы в желудке диагностировались в 12 и 16 лет. Причем у одного из них рак желудка был диагностирован в возрасте 23 лет. В семьях этих больных были указания на опухоли желудочно-кишечного тракта у родственников 1-й и 2-й степени родства, от которых они умирали в пожилом (старше 54 лет) возрасте.
Для пациентов с САП, а также для родственников отмечен высокий риск развития опухолей печени, в том числе гепатобластомы или гепатоцеллюлярного рака, которые могут возникать раньше полипов (в 2—3 года жизни). Однако, несмотря на высокий риск для пациентов САП, эти опухоли возникают редко [7, 8]. Кроме того, есть риск опухоли мозга, которые также возникают в раннем возрасте, до появления полипов. Гистологически это может быть медуллобластома (80%), пинеобластома, астроцитома или аденома (пинеалома), или кисты шишковидной железы. Важно отметить, что у носителя синдрома возможно совместное появление опухоли мозга и печени. Сочетание множественных аденом в толстой кишке с опухолью мозга известно как глиома-полипозный синдром или синдром Туркота, который также классифицирован как вариант САП.
Интересно то, что у пациентов с САП встречается ВГПЭС глаза, которая часто ассоциирует с РЩЖ [8]. РЩЖ является частым компонентом САП и поражает от 2 до 11,8% пациентов. Гистологически это, как правило, папиллярный рак, но в 1/3 случаев имеет крибриформную структуру, которая почти не встречается в общей популяции (0,2%), возникает в молодом возрасте (20 лет) и имеет благоприятный прогноз. Доброкачественные узлы в щитовидной железе встречаются у 38—79% пациентов [9].
У нашей пациентки с САП аденомы толстой кишки диагностированы в 7 лет, удалялись эндоскопически по мере их диагностики, субтотальная колэктомия с наложением асцендоректального анастомоза проведена в 14 лет. В 16 лет у нее обнаружены узлы в щитовидной железе, в 17 лет удален РЩЖ на ранней стадии. Гистологически типичный дифференцированный папиллярный рак (рис. 3). Отец больной также с синдромом САП умер от прогрессирования рака толстой кишки в 40 лет.
Рис. 3. Папиллярный рак щитовидной железы пациентки с синдромом САП. Окраска гематоксилином и эозином. ×40, ×100.
У другой пациентки РЩЖ, диагностированный в 19 лет (гистологически фолликулярный), стал первой манифестацией злокачественного фенотипа (рис. 4). Кроме того, у нее диагностирована ВГПЭС. Гемиколэктомия произведена в 27 лет, удалены умереннодифференцированная аденокарцинома и множественные полипы толстой кишки с признаками малигнизации.
Рис. 4. Фолликулярный рак щитовидной железы пациентки с синдромом САП. Окраска гематоксилином и эозином. ×40.
Для этого синдрома по сравнению с общей популяцией соотношение женщины/мужчины (17:1) много выше (3:1). РЩЖ диагностируется первым по отношению к идентификации синдрома у трети пациентов с САП, и это является причиной поиска аденом в толстой кишке у лиц с ранним развитием РЩЖ. Важным является и тот факт, что у пациентов с САП может наблюдаться косегрегация опухолей мозга и папиллярного РЩЖ [10]. Интересно наблюдение, показывающее, что пациенты с САП, ассоциированным с опухолями мозга или печени, или РЩЖ, имели ВГПЭС, и у родственников из их семей выявляли те же самые опухоли или полипы кишечника или ВГПЭС [11]. Таким образом, диагностика ВГПЭС у пациента с САП это признак, который может быть ассоциирован с появлением малигнизации не только в щитовидной железе, но и в других органах. Десмоидные опухоли обнаруживаются в семьях пациентов с САП у 25% родственников 1-й степени родства и даже у 8% — 3-й степени родства, что может быть также дополнительным признаком при идентификации синдрома.
Молекулярно-генетическая основа синдрома САП. Предрасположенность к САП вызывается герминальной мутацией в гене APC (adenomatous polyposis coli), который идентифицирован на хромосоме 5q21 в 1991 г. [12, 13]. Ген АРС относится к генам-супрессорам, состоит из 15 экзонов (причем экзон 15 занимает ¾ кодирующего сиквенса). Ген АРС кодирует протеин с молекулярной массой 311,8 кД и является основным регулятором в Wnt-сигнальном пути. Являясь частью протеинового комплекса, ген АРС участвует в регулировании фосфорилирования и деградации β-катенина. APC-протеин играет ключевую роль в адгезии эпителиальных клеток посредством связывания с Е-кадхерином и микротрубочками, регулирует миграцию клеток на кишечных криптах, пролиферацию, участвует в апоптозе, передаче внутриклеточного сигнала. Кроме того, АРС является промотором стабильности хромосом [14, 15]. Мутации в гене АРС в большинстве являются большими делециями, которые трудно выявить. Чаще всего герминальные мутации включают внутригенные инсерции, делеции, вызывающие сдвиг рамки считывания, точковые мутации, приводящие к преждевременной терминации синтеза белка и функциональной инактивации АРС-протеина. К настоящему времени известно более 900 мутаций. Что касается пациентов со слабовыраженной формой САП, то у некоторых из них выявляют мутации в гене АPС или в гене MUTYH, продукт которого участвует в эксцизионной репарации ДНК, а биаллельные мутации в этом гене приводят к развитию САП. Если мутация не выявляется ни в одном из указанных генов, одной из причин может быть большая делеция, ведущая к утрате гена. Интересно отметить, что у пациентов с САП потеря функции гена АРС начинается с герминальной потери одного аллеля гена, мутация второго аллеля зависит от места нахождения первой мутации. Было показано, что у носителей мутации в кодонах 1194—1392 вторая копия гена чаще утрачивается путем крупных делеций. Мутации вне этого региона наиболее часто точковые, которые приводят к преждевременному синтезу АРС-белка. Однако, чем ближе мутация расположена к кодону 1300, тем выше вероятность делеций и полного выключения второй копии гена, влекущего за собой приобретаемое клеткой селективное преимущество [16]. Поэтому микроскопические полипы у носителя этих мутаций неидентичные и растут с неодинаковой скоростью. Кроме того, анализ ранних аденом (3 мм) от больных САП с известными мутациями в гене АРС показал, что для дальнейшего развития туморогенеза необходимы дополнительные совместные воздействия разных факторов среды, модифицирующих генов, приобретения мутаций в генах К-ras, TP53, делеции хромосомы 18q и хромосомы 1p, влияющих на разные пути функции генов и ведущих к нарастанию хромосомной нестабильности [17].
Герминальные мутации в гене АРС являются причиной широкого спектра клинических проявлений заболевания, которые в течение жизни манифестируют в разном возрасте, и тяжесть проявления синдрома зависит от типа мутации в гене АРС. В таблице приведены данные разных авторов по локализациям мутаций в гене АРС и связанные с ними возможные фенотипические проявления заболевания (см. таблицу).
Типы мутаций в гене АРС и тяжесть проявления заболевания
Как видно из таблицы, несмотря на не очень четкие границы интервалов гена АРС между тяжелой и классической формой проявления синдрома, выявленные в гене мутации могут служить ориентиром возможной тяжести и варианта проявления фенотипа. Для аттенуированной формы САП границы интервалов в гене АРС установлены. Более легкое течение заболевания при этой форме САП объясняют сохраненной экспрессией альтернативно сплайсингового транскрипта гена АРС, не содержащего мутации, так как в его составе нет экзона 9 [21, 29]. На основании полученных корреляций можно оценить риск развития рака и планировать лечение пациентов с САП. Так, герминальная мутация, выявленная в одном из кодонов 1250—1464, предполагает более агрессивные методы лечения, особенно мутация в кодоне 1309, которая ассоциирована с ранним развитием рака толстой кишки [19].
Интересно отметить, что герминальные мутации у больных с САП и эмбриональным раком печени (гепатобластомой) в большинстве локализуются в 5’-конце кодона 1230 или кодоне 1061, а при сочетанном возникновении у пациента с САП опухоли мозга и рака печени мутация в гене АРС затрагивала тот же самый регион, как при гепатобластоме [8,11]. Кроме того, показано, что риск РЩЖ в основном обусловлен герминальной мутацией в 5’-конце экзона 15, которая считается «горячей точкой» гена АРС [8]. Однако тестирование этого гена у пациентов с САП и РЩЖ выявляло мутации в экзонах 4, 8 и 9 и было показано, что они могут встречаться на протяжении всего гена [30, 32].
Клиническая диагностика неоплазий. Риск наследования герминальной мутации потомками при этом синдроме составляет 50%. Герминальная мутация обнаруживается у 80% пациентов с полипозом, у 30% из них возникает de novo или в результате мозаицизма. Генетическое тестирование начинают с члена семьи — носителя синдрома. Других родственников в семье тестируют после идентификации мутации в гене АРС у носителя синдрома. Если мутация у пораженного полипами пациента не обнаружена в гене АРС и в гене MUTYH, родственники 1-й степени родства обследуются так же, как пациенты с САП. Если мутация обнаружена, всем родственникам, подозрительным на САП, проводится прямое тестирование ДНК, а мониторинг включает ежегодную колоноскопию с биопсией полипов, начиная с 10—12-летнего возраста, каждые 2 года вплоть до 35 лет. Для родственников колоноскопию следует начинать с пубертатного возраста или ориентируясь на такие симптомы, как диарея, боли в животе, кровь в стуле, метаболические расстройства (снижение уровней белка, холестерина, гипокалиемия), дисбактериоз, вторичный иммунодефицит. Для пациентов в старшем возрасте при первом обследовании проводится колоноскопия, затем каждые 1—2 года — сигмоидоскопия. Эндоскопию желудка, тонкой кишки рекомендовано начинать с 25—30 лет в зависимости от найденных клинических проявлений и повторять каждые 2—3 года до 50-летнего возраста. По данным литературы, у пациентов после профилактической колонэктомии рак двенадцатиперстной кишки является лидирующей причиной смерти, наиболее ранний возраст при диагностике этого рака 17 лет [33]. Полипы в области дна желудка также склонны к малигнизации. Если в кишечнике пациента полипов больше, чем можно удалить, рекомендуется колонэктомия с илеоректальным анастомозом или илеопроктоколонэктомия с последующим илеоанальным анастомозом и наблюдением за состоянием прямой кишки [34].
В семьях, где имеется один или два члена семьи со слабовыраженной формой САП, обследование начинают в более позднем подростковом возрасте (после 15 лет).
Учитывая высокий риск развития гепатобластомы у детей, анализ уровня α-фетопротеина и УЗИ-обследование брюшной полости необходимо начинать до 2 лет, повторять каждые полгода до 6 лет, у некоторых риск остается до 15-летнего возраста. Обследование головного мозга начинают после 2 лет. Следует помнить, что вышеперечисленные опухоли могут возникать до появления полипов в кишечнике. Указания на гепатобластому у одного из членов семьи являются причиной начала обследования пациента с 0,6 года с ДНК-диагностикой на носительство мутаций в гене АРС. Пальпаторное обследование щитовидной железы ежегодно, начиная с возраста 15 лет, каждые 3 года проводить УЗИ щитовидной железы. При необходимости используют тонкоигольную биопсию. Следует помнить, что у некоторых пациентов и их родственников все вышеуказанные опухоли могут появляться совместно. Тщательное ежегодное обследование щитовидной железы рекомендуется также, когда у пациента или у кого-либо из членов его семьи диагностируют ВГПЭС, или герминальную мутацию в гене АРС обнаруживают в кодонах 463−1387.
Ранней диагностике и планированию лечения пациентов с САП может помочь выявление не только внекишечных доброкачественных и злокачественных проявлений и ассоциаций между ними при этом заболевании, но и генотип-фенотипических корреляций у носителей мутаций в гене АРС.
Синдром Пейтца—Егерса (ОMIM#175200) относится к синдромам множественных гамартоматозных полипозов. Имеет аутосомно-доминантный тип наследования, распространенность 1 случай на 50 000 и 1 на 200 000 новорожденных [35]. Гамартома — это узловое образование, возникающее в результате нарушения эмбрионального развития органов и тканей, состоящее из тех же компонентов, что и орган, где оно находится, но отличается степенью дифференцировки. Клиническая картина заболевания включает одну из главных характеристик синдрома — полипы, которые могут находиться в любом отделе пищеварительного тракта. Полипы от 1 до 100 (размером от 0,1 до 3 см в диаметре), как правило, доброкачественные, развиваются у 90% пациентов, а у 1/3 пациентов выявляются к 10—13 годам. Кроме того, они могут появляться в носу, бронхах, органах репродуктивной системы, почках, уретре, мочевом и желчном пузыре.
В кишечнике полипы при гистологическом исследовании состоят из нормальных клеточных элементов пищеварительного тракта, но с измененной архитектоникой, обусловленной элонгацией пластинки эпителиального компонента слизистой оболочки кишки в строму полипа и разветвлением гладкомышечных волокон, что создает картину инвазии эпителия в толщу кишечной стенки [36, 37]. Второй характеристикой синдрома является меланиновая пигментация (от 1 до 5 мм в диаметре) коричневого или светло-коричневого цвета на границе кожи и слизистых оболочек (типично губ и щек), перианальной области, на ладонях и подошвах, слизистой кишки. Гиперпигментация возникает при рождении, может появляться в месте травмы, воспаления и бледнеть или исчезать к пубертатному периоду или с возрастом (рис. 5).
Рис. 5. Пигментации слизистой оболочки губ и полости рта при синдроме Пейтца—Егерса.
Гамартомный полипоз в кишечном тракте может сопровождаться тяжелыми осложнениями: изъязвляться, кровоточить, вызывать кишечную инвагинацию, обструкцию (обычно тонкой кишки) и даже некроз, которые в раннем детстве (до 10 лет) выявляются у 33% носителей синдрома, а к 20 годам жизни — у 50% [38]. К настоящему времени известно, что для пациентов с синдромом Пейтца—Егерса риск развития рака любой локализации в 15 раз выше, чем в общей популяции и к 65 годам жизни составляет 93% [39]. Наиболее частым местом возникновения рака являются желудочно-кишечный тракт и молочная железа. Чаще рак возникает в тонкой кишке (96%), толстой кишке (27%), желудке (24%), прямой кишке (24%) [40]. Средний возраст диагностики рака желудка 30 лет, но может возникнуть в 10 и 20 лет [39]. Риск рака молочной железы (РМЖ) такой же, как у пациентов с мутациями BRCA1 или BRCA2 (кумулятивный риск 45%). Характерно билатеральное поражение, отмечен самый ранний возраст пациента при диагностике РМЖ — 19 лет [41]. У 75% женщин возникают фиброаденомы, кисты в молочных железах, лейомиомы матки (у 44%) с высоким риском малигнизации.
Кроме того, доброкачественные заболевания щитовидной железы, включая многоузловой зоб, выявляются у 50—70% носителей синдрома, а из предшествующих фолликулярных аденом у 5 — 10% возникает фолликулярный РЩЖ [39].
Пациенты с синдромом Пейтца—Егерса с юного возраста имеют риск развития рака эндометрия, шейки матки, яичников, уретры, яичек, который чаще билатеральный и мультифокальный, пищевода, легких, поджелудочной железы. Риск возникновения рака в среднем проявляется к 42 годам [42, 43].
В нашем исследовании у пациента в возрасте 38 лет была проведена субтотальная резекция ободочной кишки по поводу первично-множественного синхронного рака. На коже в области губ и слизистых оболочек щек и перианальной области у него была выявлена пигментация, характерная для этого синдрома. Указания на постоянную диарею у его сына 7 лет стали причиной обследования и выявления небольших полипов в тощей кишке и поликистоза в почке. Удаленные полипы при гистологическом исследовании были тубулярными аденомами (рис. 6). Из других проявлений синдрома у ребенка единичные пятна гиперпигментации на пальцах. В дальнейшем в целях профилактики осложнений у ребенка неоднократно проводилось эндоскопическое удаление полипов.
Рис. 6. Тубулярная аденома. Окраска гематоксилином и эозином. ×75.
Клиническая диагностика синдрома основывается на выявлении одного из следующих проявлений заболевания:
1) 2 и более подтвержденных гистологически гамартоматозных полипов;
2) любое число полипов, выявленных у пациента, который имеет близкого родственника — носителя синдрома;
3) гиперпигментация у пациента в типичных для синдрома местах и наличие в семье родственника — носителя этого синдрома;
4) любое количество полипов в сочетании с наличием гиперпигментации в типичных местах у одного индивида [39].
Молекулярно-генетическая основа синдрома Пейтца—Егерса
Причина синдрома — герминальная мутация в гене STK11, кодирующем белок серин/треонинкиназу. Ген STK11 относится к супрессорам, картирован на хромосоме 19р13.3, включает 9 экзонов, кодирует протеин, состоящий из 433 аминокислотных остатков [44, 45]. Основные типы мутаций — небольшие делеции/инсерции, нонсенс, миссенс или большие делеции, приводящие к преждевременной терминации синтеза белка [46]. Функция гена STK11 комплексная, но до конца не установлена. Однако известно, что он регулирует клеточную пролиферацию путем ареста G1-клеточного цикла, регулирует Wnt-сигнальный путь, взаимодействуя с белком р53, участвует в апоптозе [47, 48]. Ген STK11 имеет важную роль в ориентировке клеток в пределах ткани, так как влияет на клеточную полярность и участвует в межмембранных белковых взаимодействиях [49, 50]. Протеин STK11 включается в ингибирование mTOR (mammalian target of rapamycin) пути, который известен также, как комплекс рапамицин-ассоциированных протеинов, действуя как негативный регулятор mTOR-пути [51]. Важным является то, что mTOR-путь является финальным и общим путем, который также разрегулирован при других синдромах полипозов, вызванных герминальными мутациями в генах PTEN, BMPR1A и SMAD4.
Инактивация гена STK11 является причиной развития гамартом, однако роль гамартоматозных полипов при развитии рака до конца не установлена. Обсуждается возможность малигнизации с тенденцией к последовательности: гамартома—дисплазия—рак [52]. Это предположение поддерживается находками фокусов аденом в пределах полипов, а также рака, возникающего в пределах полипа [53]. Интересными являются данные по обнаружению соматических мутаций гена STK11 в меланоме, немелкоклеточном раке легкого у пациентов — не носителей синдрома [54].
Мутации в гене STK11 выявляются у 70—80% пациентов с синдромом Пейтца—Егерса. Различия в диагностике мутаций для пациентов могут быть обусловлены методами, использованными для их выявления. Кроме того, для пациентов с фенотипом синдрома, но без выявленных мутаций в гене STK11 обсуждается гетерогенность и возможность существования второго гена, ответственного за синдром в локусе 19q13.4 [55]. Важным для мониторинга пациентов с синдромом Пейтца—Егерса является генотип-фенотипическая корреляция. Пациенты с миссенс-мутациями имели более позднее проявление симптомов заболевания, нонсенс-мутации приводят к более тяжелому течению синдрома по сравнению с другими мутациями в гене STK11 [56]. Мутации в экзоне 6 гена STK11 для пациента имеют высокий риск рака [57]. Обнаружены «горячие точки», преимущественно повреждающиеся у пациентов с герминальными мутациями в STK11, одна в кодоне 51−84 экзона 1 и также в экзоне 7. Однако до настоящего времени ясных различий между пациентами с мутациями в STK11 и теми, кто их не имел, выявлено не было [58].
Клиническая диагностика неоплазий при синдроме Пейтца—Егерса. Как и при синдроме САП, риск его наследования потомками высокий и составляет 50%. Динамическое наблюдение за пациентами — носителями синдрома преследует две главные цели: предотвратить осложнения, связанные с полипами в пищеварительном тракте (кровотечение/анемия, инвагинация и др.), и выявить рак на ранней стадии. Клиническая диагностика рака комплексная, сфокусированная на выявление полипов и неоплазий желудочно-кишечного тракта, женских репродуктивных органов, тестикулярной неоплазии. Дисплазия полипов диагностируется в раннем возрасте у 18% пациентов. Рекомендуется проводить колоноскопию, начиная с возраста 8 лет, и если полипы выявляются, эндоскопию следует проводить каждые 2—3 года совместно с наблюдением за клиническими симптомами. Если полипы не обнаружены, эндоскопическая диагностика начинается с 18 лет с 2—5-летними интервалами [58]. Эндоскопическое исследование желудка и пищевода, начиная с 10-летнего возраста, каждые 2 года. Основная тактика в эндоскопическом исследовании пищеварительного тракта — удалять все полипы более 1,5 см в диаметре [59].
Для скрининга рака молочной железы используется ежегодный врачебный осмотр, самообследование. УЗИ и маммография каждые 2—3 года, начиная с возраста 20—25 лет, после 40 лет ежегодно. Гинекологический осмотр, включая трансвагинальное УЗИ, анализ СА125, с 20-летнего возраста ежегодно. Нужно учитывать, что у мужчин может развиться опухоль яичек из клеток Сертоли (LCST), которая секретирует эстроген и приводит к увеличению молочной железы (гинекомастия) и нарушению телосложения. Поэтому регулярный осмотр яичек и УЗИ тестикул у детей и, если ничего не обнаруживается, УЗИ органов брюшной полости и тестикул начиная с 20 лет ежегодно [59].
Диагностика
Диагностируют синдром Пейтца — Егерса с помощью биопсии. Если во взятой для анализа части полипа обнаруживается гамартомная составляющая, то это типичный для данного заболевания симптом. Полипы размером от 1 до 5 мм обычно не мешают нормальному функционированию желудочно-кишечного тракта. Но по мере роста они могут привести к кровотечениям, потому наросты, размеры которых больше 1 см, необходимо удалять. Для полипов характерен умеренный рост, они могут быть как множественными, так и единичными. При множественных лечение значительно сложнее. Их нельзя удалить один раз, потому применяются щадящая диета, а также медикаментозная терапия, направленная на замедление роста новообразований.
Другие важные критерии диагностики – наследственность и пигментация слизистой. Поскольку полипы обнаруживаются у пациентов старше 10 лет, для детей пигментация на слизистых является главным признаком при постановке диагноза. Петца — Егерса синдром также довольно часто сопровождается синдромом Мак-Кьюна — Олбрайта (ранним половым развитием). Если у ребенка ранее половое развитие, вероятность развития у него гамартомного полипоза довольно высокая.
Полип желудка — симптомы и лечение
По количеству разрастаний выделяют:
- одиночные полипы;
- множественные полипы;
- полипоз желудка (более 20 полипов).
По клиническому течению некоторые советские учёные выделяли следующие формы полипов желудка:
- бессимптомная форма;
- гастритная форма;
- анемическая форма;
- осложнённая форма (кровотечение полипа и его выпадение в двенадцатиперстную кишку);
- сочетанная форма (появление полипа и рака желудка).
По эндоскопическим признакам можно выделить четыре типа полипов желудка:
- I тип — плоский, приподнятый, с нечёткими краями;
- II тип — выступающий, полукруглый, с достаточно чёткими границами;
- III тип — чётко выступающий, округлый, с втянутым основанием;
- IV тип — на ножке.
Все перечисленные классификации заслуживают внимания. Однако на практике наиболее важна классификация по признакам перерождения полипов в злокачественную опухоль [1][3].
По классификации ВОЗ, доброкачественные опухоли желудка определены как аденомы (аденоматозные полипы). Они подразделяются на папиллярные и тубулярные формы. Отдельно выделены гиперпластические полипы, которые включены в группу опухолеподобных процессов [2][10].
В 2010 году Британское общество гастроэнтерологов предложило свою классификацию полипов желудка, а также выработало рекомендации по ведению пациента при каждом типе полипов желудка. Согласно этой классификации, полипы желудка делят на пять групп:
- Полипы фундальных желёз.
- Гиперпластический полип.
- Аденоматозный полип.
- Гамартомные полипы (ассоциированные с пороками развития):
- ювенильный полип;
- синдром Пейтца — Егерса;
- синдром Коудена.
- Полипозные синдромы (негамартомные):
- ювенильный полипоз;
- семейный аденоматозный полипоз.
Полипы фундальных желёз — это кистозные расширения собственных желёз желудка, составляют 16-51 % доброкачественных полипов. В диаметре обычно достигают 1-5 мм, располагаются в основном в теле или дне желудка. Имеют гладкую ровную поверхность, могут быть дольчатыми, покрыты неизменённой слизистой оболочкой. Могут появляться как самостоятельное заболевание или в составе семейного аденоматозного полипоза толстой кишки.
Не связаны с гастритом и хеликобактерной инфекцией. Могут образоваться на фоне длительного приёма ингибиторов протонной помпы (препаратов, снижающих выработку соляной кислоты). Эти лекарства повышают активность гастрина (гормона желудка), который стимулирует рост эпителиальных клеток.
Средний интервал развития полипов фундальных желёз — 32,5 месяца. Регресс наступает через три месяца после прекращения приёма ингибиторов протонной помпы [4][7].
Гиперпластический полип составляет 30-93 % доброкачественных полипов желудка. Может быть сидячим и на ножке, в диаметре менее 2 см. Отличается увеличением желудочных ямок, расширенными и извилистыми железами, хроническим воспалением слизистой оболочки желудка. Единичный полип чаще всего располагается в антральном (нижнем) отделе желудка. Множественные полипы могут возникать во всех отделах желудка.
Связан с хроническим (хеликобактер-ассоциированным), химическим и атрофическим гастритом. Возникает из-за повышенного обновления клеток (их наслаивания друг на друга) в ответ на повреждение эпителия желудка (обычно при эрозиях или язве желудка).
Сам по себе гиперпластический полип редко становится злокачественным (малигнизуется), однако он повышает риск малигнизации окружающей воспалённой ткани желудка. Поэтому при обнаружении гиперпластического полипа рекомендуется выполнить биопсию окружающей ткани (из 4-5 разных мест) [4][5].
Аденоматозный полип — это предраковое заболевание с высоким потенциалом перерождения в рак, особенно при полипе более 2 см. Составляет 3-26 % полипов желудка. Обычно одиночный, может локализоваться в любом отделе желудка, но чаще обнаруживается в антруме (нижней части желудка). По строению бывает трубчатым, ворсинчатым и смешанным. Возникает на фоне атрофического гастрита и кишечной метаплазии (когда желудочный эпителий заменяется кишечным) [4].
Стоит отметить, что в одном исследовании в Витебской областной клинической больнице обнаружили гиперпластический полип с участками аденоматоза. Авторы предположили, что гиперпластические полипы могут трансформироваться в аденоматозные. Они также позволили себе выделить ещё одну гистологическую форму полипов — гиперпластический полип с очаговым аденоматозом. Он представляет собой истинную доброкачественную опухоль желудка, способную к перерастанию в рак [10].
Гамартомные полипы встречаются редко, однако пару слов о них сказать стоит.
Одиночный ювенильный (юношеский) полип не имеет злокачественного потенциала, но, как и все полипы, может осложниться кровотечением или ущемлением, так как в основном такие полипы располагаются в нижней части желудка и подвержены травматизации.
Синдром Пейтца — Егерса — редкое наследственное заболевание, которое сопровождается появлением гамартомных полипов в желудочно-кишечном тракте, а также пигментацией в области губ, пальцев и слизистой оболочки щёк. При данном заболевании высок риск малигнизации как органов пищеварения, так и лёгких, молочных желёз, поджелудочной железы, матки. Поэтому такие пациенты должны находиться под динамическим наблюдением.
Синдром Коудена — редкое наследственное заболевание, которое сопровождается наличием полипов желудочно-кишечного тракта, доброкачественных опухолей области рта, а также пороков развития различных органов (молочных желёз, щитовидной железы и гениталий). Полипы при данном синдроме перерождаются в рак очень редко, но всё же требуют наблюдения.
Полипозные синдромы включают в себя ювенильный полипоз и семейный полипозный синдром.
При ювенильном полипозе обнаруживают множество ювенильных полипов, которые имеют злокачественный потенциал. Также это заболевание может осложняться желудочно-кишечным кровотечением и энтеропатией — заболеванием тонкой кишки, которое сопровождается потерей белка и других питательных веществ.
Семейный полипозный синдром является наследственно-опосредованным заболеванием с очень высоким риском рака желудка и других отделов желудочно-кишечного тракта. У поражённых членов семьи имеется большое количество толстокишечных и ректальных аденом, которые с вероятностью 100 % перерастают в рак, если не провести профилактическую колэктомию — полное удаление толстой кишки. Полипы желудка выявляются в 30-100 % случаев. При данном синдроме нет чёткой связи с гастритом, вызванным хеликобактерией [4].
Конкретных стадий развития заболеваний не выделяют, так как разные виды полипов имеют различное происхождение и развитие. Но если говорить о часто встречающихся аденоматозных и гиперпластических полипах, то условно можно выделить три стадии развития [1]:
- полипозный гастрит (воспаление или атрофия слизистой оболочки желудка);
- полип желудка;
- рак желудка.
Синдром Пейтса-Егерса
«Синдром Пейтца-Йегерса»
— генетическое исследование для выявления патогенных мутаций в гене STK11 ассоциированном с риском развития синдрома Синдром Пейтца-Йегерса. Панель разработана на основании клинических рекомендаций Американской ассоциации онкологов версия 1.2019. Синдром Пейтца-Йегерса – заболевание с аутосомно-доминантным типом наследования (то есть для его развития достаточно наличия мутации у одного из родителей), характеризуется развитием доброкачественных новообразований, называемых гамартоматозными полипами, в желудочно-кишечном тракте (особенно в желудке и кишечнике) и значительно повышенным риском развития некоторых типов рака, а также характерных пигментированных участков на коже и слизистых. У большинства пациентов они появляются в течение первого года жизни и часто исчезают по мере взросления человека. Полипы могут вызывать проблемы со здоровьем, такие как периодическая непроходимость кишечника, хроническое кровотечение и боль в животе. Большинство случаев данного заболевания вызваны мутацией в гене STK11 (LKB1).
Белок STK11 (серин-треониновая киназа 11) снижает скорость слишком быстрого роста и деления клеток, участвует в инициации апоптоза (активирует запрограммированную гибель клетки). Мутации в гене ассоциированы с неконтролируемым ростом и делением клеток, что приводит к образованию доброкачественных полипов и раковых опухолей.
Исследование включает определение 14 полиморфизмов гена STK11
1. STK11 Серин-треониновая киназа 11 (g.1221237C>A; c.759C>A; p.Tyr253Ter; rs137853075) 2. STK11 Серин-треониновая киназа 11 (g.1221321del; c.843del; p.Leu282SerfsTer5; rs587776656) 3. STK11 Серин-треониновая киназа 11 (g.1220701_1220704del; c.718_721del; p.Ser240LeufsTer46; rs587776657) 4. STK11 Серин-треониновая киназа 11 (g.1220372G>A; c.465-1G>A; rs587776658) 5. STK11 Серин-треониновая киназа 11 (g.1207163A>T/G; c.250A>T/G; p.Lys84Ter/Glu; rs137853076) 6. STK11 Серин-треониновая киназа 11 (g.1221312_1221313del; c.834_835del; p.Cys278TrpfsTer6; rs587776659) 7. STK11 Серин-треониновая киназа 11 (g.1207113T>C; c.200T>C; p.Leu67Pro; rs137853077) 8. STK11 Серин-треониновая киназа 11 (g.1221994_1222002del; c.908_916del; p.Ile303_His306delinsAsn; rs587776660) 9. STK11 Серин-треониновая киназа 11 (g.1207082G>T; c.169G>T; p.Glu57Ter; rs137854584) 10. STK11 Серин-треониновая киназа 11 (g.1219367del; c.418del; p.Leu140TrpfsTer21; rs397518440) 11. STK11 Серин-треониновая киназа 11 (g.1207110dup; c.197dup; p.Leu67AlafsTer96; rs397518441) 12. STK11 Серин-треониновая киназа 11 (g.1220700G>C; c.717G>C; p.Trp239Cys; rs137853082) 13. STK11 Серин-треониновая киназа 11 (g.1221977del; c.891del; p.Arg297SerfsTer39; rs587776661) 14. STK11 Серин-треониновая киназа 11 (g.1221216C>G; c.738C>G; p.Tyr246Ter; rs137853083)
Показания
Определение генетической предрасположенности к развитию злокачественных опухолей и диагностики синдрома Пейтца-Йегерса. А также для раннего выявления мутаций у родственников пациентов с данным синдромом.
Подготовка
Взятие образцов венозной крови для исследования ДНК может производиться в любое время суток натощак (8-12 часов, но не более 14 часов) или не ранее, чем через 4 часа после необильного приема пищи, специальная диета и приём лекарственных препаратов не повлияют на результат. Допустимо пить чистую воду без газа и сахара. Накануне избегать пищевых перегрузок. Исключить прием алкоголя не менее чем за 24 часа до взятия крови.
Обращаем внимание, что на результаты исследования ДНК могут повлиять переливание крови или трансплантация костного мозга, которые проводились в недавнем времени (в данном случае исследование ДНК не рекомендуется проводить в течение, как минимум, 1 месяца после трансплантации или переливания крови).
Интерпретация результатов
Для интерпретации результатов генетического тестирования требуется консультация врача-генетика.