1.What is esophageal spasm?
Esophageal spasm
- These are irregular, involuntary and sometimes very strong contractions of the esophagus - the tube through which food passes from the mouth to the stomach.
In general, the esophagus normally contracts to move food, but a spasm is a little different. There are two types of esophageal spasm:
- Diffuse spasm of the esophagus (esophagospasm)
. This is an irregular and uncoordinated contraction of the muscles of the esophagus. Such a spasm may prevent food from entering the stomach, and it gets stuck in the esophagus. - The second type
is a spasm, in which the spasm contracts using the same mechanism as during normal movement of food. These contractions allow food to move through the esophagus, but can cause severe pain.
Both types of esophageal spasms can occur in the same person.
In general, esophageal spasm is a rare problem. Often symptoms that may be mistaken for esophageal spasm are the result of other problems.
– GERD (gastroesophageal reflux disease), achalasia, problems with the nervous system in which the muscles of the esophagus and lower esophageal sphincter do not work as they should. Panic attacks can also cause symptoms similar to esophageal spasms.
A must read! Help with treatment and hospitalization!
2.Causes of spasms
The exact causes of esophageal spasm are not known. Many doctors believe that esophageal spasm is the result of a disturbance in the nervous activity that coordinates the swallowing movements of the esophagus. Some people may experience cramping due to very hot and very cold foods. Microtrauma due to solid food, infectious diseases such as measles, scarlet fever or influenza, stress, inflammatory diseases of the esophageal mucosa (esophagitis, gastric and duodenal ulcers) are also considered possible causes of the development of esophageal spasm.
Visit our Gastroenterology page
Signs of disease development
The level of development of the pathology is diagnosed endoscopically. There are several degrees of development of the disease.
- First degree. The mobile cardia does not close completely. In this case, the open gap should not be more than 1/3 of the total diameter during deep breathing. Among the external manifestations are frequent belching of air.
- Second degree. The cardiac sphincter closes only halfway. The gaping may even be slightly excessive. Belching becomes painful and very frequent. Sometimes prolapse of the gastric mucosa develops.
- Third degree. The cardiac sphincter does not close at all. There are signs of esophagitis.
The main symptoms of mild cardia include the following:
- heartburn that appears at different times relative to food intake;
- burning pain that spreads behind the sternum, where the esophagus passes;
- frequent belching with air, as well as with what is in the stomach (if the belching is sour, it means that it contains only the contents of the stomach, if there is a bitter taste, it means that bile has been added);
- nausea, which often ends with vomiting;
- esophagitis, which appears due to irritation of the mucous membrane, gives abdominal pain, and there is murmur in the intestines;
- weakness, dizziness, fatigue.
3.Diagnostics
Esophageal spasm can often be diagnosed by reviewing the patient's medical history. The doctor may ask you several questions about what foods or liquids caused the discomfort, where these symptoms are felt and how, whether you feel like food is getting stuck in the esophagus, and some others.
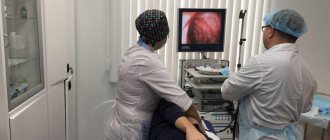
The diagnosis of esophageal spasm can be confirmed using special tests, including esophageal manometry
.
Esophageal manometry measures pressure. The second study is an x-ray with a preliminary swallow of barium solution,
which allows you to visually examine the esophagus.
Other specific tests may be performed to determine if symptoms are due to other conditions - GERD, reverse reflux of food and stomach acids, etc.
About our clinic Chistye Prudy metro station Medintercom page!
Modern approaches to the treatment of patients with hypertrophic cardiomyopathy
The half-century history of studying the problem of hypertrophic cardiomyopathy (HCM) reflects a significant evolution of knowledge in the field of etiology, pathogenesis, diagnosis, clinical course, prognosis and treatment options for this disease. During this period, more than 1,000 major scientific works were published in English-language publications alone. HCM is one of the main and probably the most common forms of cardiomyopathies, diseases of the myocardium accompanied by its dysfunction (Report of the 1995 WHO/ISFC Task Force on the Definition and Classification of Cardiomyopathy) [1].
In 2003, an International Committee (ACC/ESC) was created, bringing together American and European experts on HCM, and a report was published summarizing the main provisions, including the treatment strategy [2].
The definition of the disease is descriptive. Thus, according to modern concepts, HCM is a predominantly genetically determined disease of the heart muscle, characterized by a complex of specific morphofunctional changes and a steadily progressive course with a high threat of developing severe, life-threatening arrhythmias and sudden death (SD). HCM is characterized by massive hypertrophy of the myocardium of the left and/or less often the right ventricle, often asymmetrical in nature due to thickening of the interventricular septum (IVS), often with the development of obstruction (systolic pressure gradient) of the LV outflow tract in the absence of visible causes (arterial hypertension, defects and specific heart diseases ). The main diagnostic method remains echocardiographic examination. Depending on the presence or absence of a systolic pressure gradient in the LV cavity, HCM is divided into obstructive and non-obstructive, which is of great practical importance when choosing treatment tactics. In this case, 3 hemodynamic variants of obstructive HCM are distinguished: with subaortic obstruction at rest (the so-called basal obstruction); with labile obstruction, characterized by significant spontaneous fluctuations in the intraventricular pressure gradient for no apparent reason; with latent obstruction, which is caused only by exercise and provocative pharmacological tests (in particular, inhalation of amyl nitrite, when taking nitrates or intravenous administration of isoproterenol).
Typical morphological changes are: anomaly in the architectonics of the contractile elements of the myocardium (hypertrophy and disorientation of muscle fibers), the development of fibrotic changes in the heart muscle, and pathology of small intramyocardial vessels [3, 4].
Symptoms of the disease are varied and non-specific, associated with hemodynamic disorders (diastolic dysfunction, dynamic obstruction of the outflow tract, mitral regurgitation), myocardial ischemia, pathology of autonomic regulation of blood circulation and disruption of electrophysiological processes in the heart [5, 6, 7]. The range of clinical manifestations is extremely wide: from asymptomatic to steadily progressing forms that are difficult to treat with medication, accompanied by severe symptoms. In this case, the first and only manifestation of the disease may be sudden death.
Currently, there is a widespread increase in the number of registered cases of this pathology, both due to the introduction of modern diagnostic methods into practice, and, probably, due to a true increase in the number of patients with HCM [8, 9]. According to recent studies, the prevalence of the disease in the general population is higher than previously thought, amounting to 0.2% [10, 11]. HCM can be diagnosed at any age, from the first days to the last decade of life, but the disease is predominantly detected in people of young working age [12, 13]. The annual mortality rate of patients with HCM ranges from 1 to 6%: in adult patients it is 1–3% [14, 15], and in childhood and adolescence in people at high risk of VS it is 4–6% [16, 17].
The concept of the predominantly hereditary nature of HCM is generally accepted [18, 19]. The term “familial hypertrophic cardiomyopathy” is widely used in the literature. To date, it has been established that more than half of all cases of the disease are inherited [20, 21], with the main type of inheritance being autosomal dominant. The rest fall on the so-called sporadic form; in this case, the patient does not have relatives with HCM or myocardial hypertrophy. Most, if not all cases of sporadic HCM are also thought to have a genetic cause, meaning they are caused by random mutations.
HCM is a genetically heterogeneous disease caused by more than 200 described mutations in several genes encoding myofibrillar proteins [2, 22]. To date, 10 protein components of the cardiac sarcomere are known that perform contractile, structural or regulatory functions, defects of which are observed in HCM. Moreover, in each gene, many mutations can cause a disease (polygenic multiallelic disease).
The presence of one or another mutation associated with HCM is recognized as the “gold” standard for diagnosing the disease. Moreover, the described genetic defects are characterized by varying degrees of penetrance and the severity of morphological and clinical manifestations. The severity of the clinical picture depends on the presence and degree of hypertrophy. Mutations that are associated with high penetrance and poor prognosis are associated with greater left ventricular hypertrophy and IVS thickness than those with low penetrance and good prognosis. Thus, it has been shown that only certain mutations are associated with a poor prognosis and a high incidence of VS. These include substitutions Arg403Gln, Arg453Cys, Arg719Trp, Arg719Gln, Arg249Gln in the β-myosin heavy chain gene, InsG791 in the myosin-binding protein C gene, and Asp175Asn in the α-tropomyosin gene [23, 24, 25, 26]. Mutations in the troponin T gene are characterized by moderate myocardial hypertrophy, but the prognosis is quite unfavorable, and the likelihood of sudden cardiac arrest is high [27]. Other genetic anomalies, as a rule, are accompanied by a benign course and a favorable prognosis, or occupy an intermediate position in the severity of the manifestations they cause.
Thus, HCM is characterized by extreme heterogeneity of its causes, morphological, hemodynamic and clinical manifestations, a variety of course and prognosis options, which significantly complicates the choice of adequate and most effective therapeutic approaches to control and correct existing disorders. In this case, 5 main options for the course of the disease and outcomes are clearly distinguished:
- stable, benign course;
- sudden death;
- progressive course - increased shortness of breath, weakness, fatigue, pain (atypical pain, angina), the appearance of syncope, disorders of LV systolic dysfunction;
- “final stage” - further progression of congestive heart failure associated with LV remodeling and systolic dysfunction;
- development of atrial fibrillation and associated complications, in particular thromboembolic ones.
The variability of the prognosis determines the need for detailed stratification of the risk of fatal complications of the disease, the search for available prognostic predictors and criteria for evaluating the treatment.
According to modern concepts, the treatment strategy is determined by dividing patients into categories depending on the course and prognosis options described above (Fig.).
All individuals with HCM, including carriers of pathological mutations without phenotypic manifestations of the disease and patients with asymptomatic disease, require dynamic monitoring, during which the nature and severity of morphological and hemodynamic disorders are assessed. Of particular importance is the identification of factors that determine an unfavorable prognosis and an increased risk of VS (in particular, hidden prognostically significant arrhythmias).
General measures include limiting significant physical activity and prohibiting sports that can worsen myocardial hypertrophy, increase the intraventricular pressure gradient and the risk of VS. To prevent infective endocarditis in situations associated with the development of bacteremia, with obstructive forms of HCM, antibiotic prophylaxis is recommended, similar to that in patients with heart defects.
To date, the issue of the need for active drug therapy in the largest group of patients with asymptomatic or oligosymptomatic forms of HCM and a low probability of VS has not been finally resolved. Opponents of active tactics point out that with a favorable course of the disease, life expectancy and mortality rates do not differ from those in the general population [28, 29]. Some authors indicate that the use of β-blockers and calcium antagonists (verapamil) in this group of patients can lead to inhibition of hemodynamic disorders and clinical symptoms [30, 31]. At the same time, no one disputes the fact that watchful waiting in cases of asymptomatic or low-symptomatic HCM is possible only in the absence of signs of intraventricular obstruction, fainting and serious cardiac arrhythmias, complicated heredity and cases of VS in close relatives.
It should be recognized that the treatment of HCM, a genetically determined disease usually recognized at a late stage, may still be largely symptomatic and palliative. Nevertheless, the main objectives of therapeutic measures include not only the prevention and correction of the main clinical manifestations of the disease with improving the quality of life of patients, but also a positive effect on the prognosis, prevention of cases of VS and disease progression.
The basis of drug therapy for HCM is drugs with a negative inotropic effect: β-blockers and calcium channel blockers (verapamil). Disopyramide and amiodarone are also used to treat cardiac arrhythmias, which are very common in this disease.
β-blockers became the first and remain to this day the most effective group of drugs used in the treatment of HCM. They have a good symptomatic effect in relation to the main clinical manifestations: shortness of breath and palpitations, pain syndrome, including angina pectoris, in at least half of patients with HCM [32, 33, 34], which is mainly due to the ability of these drugs to reduce myocardial oxygen demand . Due to the negative inotropic effect and reduction of activation of the sympathoadrenal system during physical and emotional stress, β-blockers prevent the emergence or increase of the subaortic pressure gradient in patients with latent and labile obstruction, without significantly affecting the magnitude of this gradient at rest. The ability of β-blockers to improve the functional status of patients during a course and long-term use has been convincingly demonstrated [35]. Although the drugs do not directly affect myocardial diastolic relaxation, they may improve LV filling indirectly by reducing heart rate and preventing myocardial ischemia [36]. There is evidence in the literature confirming the ability of β-blockers to inhibit and even reverse the development of myocardial hypertrophy [37, 38]. However, other authors emphasize that the symptomatic improvement caused by β-blockers is not accompanied by regression of LV hypertrophy and improvement in patient survival [39]. Although the effect of these drugs in relieving and preventing ventricular and supraventricular arrhythmias and sudden death has not been proven, a number of experts still consider it advisable to prescribe them prophylactically to patients with high-risk HCM, including young patients with a family history of sudden death [40].
Preference is given to β-blockers without intrinsic sympathomimetic activity. The greatest experience has been accumulated in the use of propranolol (obzidan, anaprilin). It is prescribed starting at 20 mg 3-4 times a day, with a gradual increase in dose under the control of pulse and blood pressure (BP) to a maximum tolerated in most cases of 120-240 mg/day. One should strive to use the highest possible doses of the drug, since the lack of effect of beta-blocker therapy is likely due to insufficient dosage. However, we must not forget that increasing dosages significantly increases the risk of known side effects.
Currently, the possibility of effective use of a new generation of long-acting cardioselective β-blockers, in particular atenolol, Concor, etc., is being widely studied. However, there is an opinion that cardioselective β-blockers in patients with HCM do not have advantages over non-selective ones, since in large cases doses, which should be achieved, selectivity is practically lost. It should be noted that sotalol, recommended for use in patients with HCM with severe supraventricular and ventricular arrhythmias, combines the properties of non-selective β-blockers and class III antiarrhythmic drugs (cordarone-like effect).
The use of slow calcium channel blockers in HCM is based on reducing the level of free calcium in cardiomyocytes and leveling the asynchrony of their contraction, improving myocardial relaxation and reducing its contractility, and suppressing the processes of myocardial hypertrophy. Among calcium channel blockers, the drug of choice, due to the greatest severity of the negative inotropic effect and the most optimal profile of pharmacological properties, is verapamil (isoptin, finoptin). It provides a symptomatic effect in 65–80% of patients, including cases of refractory treatment with β-blockers, which is due to the drug’s ability to reduce myocardial ischemia, including painless, and improve diastolic relaxation and LV compliance [41, 42, 43]. This property of verapamil provides an increase in patients' tolerance to physical activity and a decrease in the subaortic pressure gradient at rest, with a lower ability to reduce intraventricular obstruction in cases of physical and emotional stress and provocation with isoproterenol compared to β-blockers. At the same time, verapamil reduces peripheral vascular resistance due to its vasodilator effect [44]. And although this effect is most often offset by a direct positive effect on LV diastolic function, in some patients with basal intraventricular obstruction in combination with increased LV end-diastolic pressure and a tendency to systemic arterial hypotension with a decrease in afterload, the intraventricular pressure gradient can increase sharply. This can lead to the development of pulmonary edema, cardiogenic shock, and even sudden death [45]. Similar serious complications of pharmacotherapy with verapamil have also been described in patients with non-obstructive HCM with high pressure in the left atrium, in whom they are caused by the negative inotropic effect of the drug. It is obvious how important it is to be careful when starting treatment with verapamil in this category of patients. The drug should be started in a hospital setting with small doses - 20-40 mg 3 times a day with a gradual increase if tolerated well until the resting heart rate decreases to 50-60 beats/min. The clinical effect usually occurs when taking at least 160–240 mg of the drug per day; Prolonged forms (isoptin retard, verogalide retard) are more convenient for long-term use. Given the beneficial effects of verapamil on diastolic function and the magnitude of the subaortic pressure gradient in the LV, as well as the proven ability to increase the survival of patients with HCM compared with placebo [46], its prophylactic administration in asymptomatic patients with high-risk HCM is advisable.
The place of diltiazem in the treatment of HCM has not been fully determined. There is evidence that at an average dose of 180 mg/day for 3 doses, it has the same pronounced beneficial effect on LV diastolic filling as 240 mg verapamil and the same symptomatic effect, but to a lesser extent improves the physical performance of patients [47].
Our clinic continues prospective observation (from 1 to 5 years) of more than 100 patients with HCM. Patients were randomized into 3 groups comparable in number, gender, age and severity of clinical manifestations. Patients were randomly assigned to atenolol or isoptin retard; in the third group, people with severe ventricular arrhythmias predominated, and they were recommended to take sotalol. The effectiveness of various drug treatment options was assessed under conditions of long-term (at least 1 year) use of drugs. Daily doses for a double dose regimen were titrated individually and averaged 85, 187, 273 mg for atenolol, isoptin and sotalol, respectively. Long-term therapy led to an improvement in the clinical condition in 77, 72 and 83% of patients in each group, respectively, which was expressed in a significant decrease in the main symptoms, manifestations of heart failure (HF), an increase in power and time of exercise, and an improvement in quality of life indicators (25, 32 and 34% respectively). At the same time, a significant (p < 0.01) decrease in heart rate at rest and at the peak of physical activity, positive dynamics of myocardial perfusion and diastolic function indicators were revealed. No significant changes in the main echocardiographic parameters were recorded during treatment; a tendency to a decrease in the pressure gradient in the left ventricular outflow tract in patients with obstructive HCM was noted. At the same time, in patients with initially severe disturbances of LV diastolic function (restrictive and “pseudonormal” type of transmitral flow), therapy was ineffective.
Thus, long-term therapy with atenolol, isoptin and sotalol has a similar beneficial effect on the condition of the majority of patients with HCM: it reduces the clinical manifestations of the disease, improves the functional state and quality of life, which is associated with their positive effect on hemodynamics and myocardial perfusion [48, 49].
It should be noted that β-blockers (with the exception of sotalol) and calcium antagonists have weak antiarrhythmic activity, while the frequency of dangerous ventricular and supraventricular arrhythmias in patients with HCM is extremely high. Therefore, it is important to use antiarrhythmic drugs in this category of patients, among which the most popular and recommended by leading experts is disopyramide.
Disopyramide (rhythmilen), a class IA antiarrhythmic, has a pronounced negative inotropic effect; in patients with HCM it can reduce the level of obstruction of the LV outflow tract and has a positive effect on the structure of diastole. The effectiveness of long-term treatment with disopyramide has been proven in patients with HCM with moderate LV outflow obstruction [50]. It is especially beneficial to use this drug in patients with ventricular arrhythmias. The initial dose is usually 400 mg/day with a gradual increase to 800 mg. In this case, as in the case of prescribing sotalol, it is necessary to monitor the duration of the QT interval using an ECG.
An equally effective treatment and prevention of both ventricular and supraventricular arrhythmias in HCM is amiodarone (cordarone), which, along with antiarrhythmic activity, apparently somewhat reduces hypercontractility and myocardial ischemia. Moreover, according to W. McKenna et al. [51], its ability to prevent sudden death in such patients has been shown. Treatment with amiodarone begins with saturating doses (600–1200 mg/day) for 3–7 days with a gradual reduction to maintenance doses (preferably 200 mg/day or less) as the heart rate decreases. Considering the deposition of the drug in tissues with possible dysfunction of the thyroid gland, the development of pneumofibrosis, damage to the cornea, skin and liver, with its long-term (more than 10-12 months) use, regular monitoring of the condition of these “vulnerable” organs is necessary in order to early identify possible complications of pharmacotherapy .
In HCM, combinations of drugs with a negative inotropic effect are possible, for example, β-blockers and calcium antagonists, β-blockers and disopyramide.
Signs of venous congestion in the lungs, including nocturnal attacks of cardiac asthma, are not uncommon in HCM and in most cases are due to LV diastolic dysfunction. Such patients are treated with β-blockers or calcium antagonists in combination with careful use of saluretics. Peripheral vasodilators, including nitrates, and cardiac glycosides should be avoided due to the risk of worsening LV diastolic filling and a sharp decrease in cardiac output, including the development of syncope and sudden death.
Various supraventricular tachyarrhythmias, mainly atrial fibrillation and flutter, are observed in 10–30% of patients with HCM [52, 53] and pose a risk of occurrence or aggravation of cardiohemodynamic disorders, the occurrence of thromboembolism, as well as an increased risk of developing ventricular fibrillation due to frequent concomitant atrioventricular dysfunction. connections and the presence of additional pathways between the atria and ventricles. As a result, in patients with HCM, paroxysmal supraventricular arrhythmias are classified as potentially fatal, and the rapid restoration of sinus rhythm and the prevention of repeated paroxysms become especially important.
To relieve paroxysms of atrial fibrillation, in addition to group IA antiarrhythmic drugs and amiodarone, β-blockers, verapamil and digoxin are used; if they are ineffective, electrical pulse therapy is used [54]. For persistent atrial fibrillation, β-blockers or verapamil in combination with digoxin are used to control the ventricular rate. This is the only case when patients with obstructive HCM can be prescribed cardiac glycosides without fear of increasing the intraventricular pressure gradient. Since atrial fibrillation in patients with HCM is associated with a high risk of systemic thromboembolism, including stroke, immediately after its development it is necessary to begin therapy with anticoagulants, which, with a permanent form of atrial fibrillation, are taken indefinitely [55].
Unfortunately, in a significant number of patients with HCM, traditional pharmacotherapy does not effectively control the symptoms of the disease; patients are not satisfied with the low quality of life. In such cases, it is necessary to decide on the possibility of using other, non-drug treatment approaches. In this case, further tactics are determined separately in patients with obstructive and non-obstructive forms of HCM.
Contrary to popular belief, often at an advanced stage of the pathological process (mainly in the non-obstructive form of HCM), progressive systolic dysfunction and severe heart failure develop, associated with LV remodeling (thinning of its walls and dilatation of the cavity). This evolution of the disease is observed in 2–5% of patients with HCM and characterizes the final (“dilated”) stage of a special, severe and accelerated process that does not depend on the patient’s age and how long ago the disease has manifested itself [56, 57]. The increase in LV systolic size usually precedes and prevails over diastolic expansion. The clinical features of this stage are severe, often refractory congestive heart failure and an exceptionally poor prognosis. The treatment strategy for such patients is changing and is based on the general principles of treatment of congestive heart failure, and involves careful prescription of ACE inhibitors and angiotensin II receptor blockers, diuretics and cardiac glycosides, β-blockers and spironolactone. These patients are candidates for heart transplantation.
In the absence of clinical effect from active drug therapy in symptomatic patients of functional class III-IV according to the classification of the New York Heart Association with severe asymmetric IVS hypertrophy and a subaortic pressure gradient at rest equal to 50 mm Hg. Art. and more, surgical treatment is indicated [58, 59]. The classic technique is transortic septal myectomy, proposed by Agmorrow. In young patients with a family history of HCM with severe clinical manifestations and indications of early VS in relatives, the indications for the patient should be expanded [60]. In some centers it is also performed in cases of significant latent obstruction. In general, at least 5% of all patients with HCM are potential candidates for surgical treatment. The operation provides a good symptomatic effect with complete elimination or significant reduction of the intraventricular pressure gradient in 95% of patients and a significant reduction in LV end-diastolic pressure in 66% of patients [61, 62]. Surgical mortality is currently around 1–2%, which is comparable to the annual mortality rate with medical therapy (2–5%) [2]. Although most earlier studies failed to find a significant effect of surgical treatment of HCM on prognosis, Seiler et al. [63] showed an improvement in the 10-year survival rate of operated patients to 84% compared to 67% in the group treated with medication. There are reports of 40-year follow-up after myectomy.
In some cases, if there are additional indications to reduce the severity of obstruction and mitral regurgitation, valvuloplasty or mitral valve replacement with a low-profile prosthesis is performed simultaneously. Long-term results of myectomy can be improved by subsequent long-term therapy with verapamil, which provides improvement in LV diastolic function, which is not achieved with surgical treatment.
Currently, techniques different from classical myectomy have been developed and are successfully used. In particular, at the Scientific Center for Agricultural Sciences named after. A. N. Bakulev, under the leadership of Academician L. A. Bockeria, developed an original technique for excision of the zone of hypertrophied IVS from the conical part of the right ventricle. This method of surgical correction of obstructive HCM is highly effective and can be the method of choice in cases of simultaneous obstruction of the outflow tracts of both ventricles, as well as in cases of midventricular LV obstruction.
In recent years, there has been growing interest in studying the possibility of using sequential dual-chamber pacing with a shortened atrioventricular delay as an alternative to surgical treatment of patients with obstructive HCM [64]. The resulting change in the sequence of propagation of the wave of excitation and contraction of the ventricles, which first covers the apex and then the IVS, leads to a decrease in the subaortic gradient due to a decrease in regional contractility of the IVS and, as a consequence, expansion of the LV outflow tract. This is also facilitated by a delay in the anterior systolic movement of the anterior MV leaflet and a decrease in its amplitude. It is important to select the shortest delay time for the application of the ventricular impulse after the atrial impulse, which ensures premature depolarization of the apex of the heart without leading to a deterioration in cardiohemodynamics—a decrease in cardiac output and blood pressure. To achieve this, in some cases it is necessary to resort to lengthening the time of spontaneous atrioventricular conduction using therapy with β-blockers or verapamil and even ablation of the atrioventricular node. Although initial uncontrolled observations were very encouraging, more recent randomized trials have shown that the symptomatic effect achieved by pacing and the reduction in subaortic pressure gradient (about 25%) are relatively small, and there are no significant changes in physical performance at all [65, 66]. It was not possible to detect a significant effect of cardiac pacing on the incidence of sudden death. Concerns are caused by worsening impaired myocardial diastolic relaxation and increased LV end-diastolic pressure. It is obvious that until the role of cardiac pacing in the treatment of obstructive HCM is clarified, extended clinical use of this method is not recommended.
Another alternative treatment for refractory obstructive HCM is transcatheter alcohol septal ablation [67, 68]. The technique involves infusion of 1–3 ml of 95% alcohol through a balloon catheter into the perforating septal branch, resulting in an infarction of the hypertrophied section of the IVS, involving from 3 to 10% of the mass of the LV myocardium (up to 20% of the mass of the IVS). This leads to a significant reduction in the severity of outflow tract obstruction and mitral insufficiency, objective and subjective symptoms of the disease [69, 70]. Moreover, in 5–10% of cases there is a need to implant a permanent pacemaker due to the development of high-degree atrioventricular block. In addition, to date, the positive effect of transcatheter ablation on prognosis has not been proven, and operative mortality (1-2%) does not differ from that during septal myectomy, which is currently considered the “gold standard” for the treatment of patients with HCM with severe symptoms and obstruction LV outflow tract resistant to pharmacotherapy.
Risk stratification of sudden death in patients with HCM
According to most authors, the undeniable factors of high risk of VS in HCM are: young age (<14 years); patients have a history of fainting and severe ventricular arrhythmias, episodes of unstable ventricular tachycardia according to the results of daily ECG monitoring; inappropriate increase in blood pressure during the stress test; pronounced (more than 3 cm) hypertrophy of the LV myocardium; indication of HCM and/or sudden death in the family history [71, 72]. In addition, some researchers believe that the likelihood of VS increases if the patient has atrial fibrillation, severe myocardial ischemia and LV outflow tract obstruction [73]. Great importance is attached to the detection of mutations associated with a severe prognosis in patients with a familial nature of the disease. Establishing a high risk of VS determines the need for special, more active medical tactics in relation to this category of patients (clarification of drug therapy, use of pacemakers, defibrillators-cardioverters, surgical interventions). In this case, the most adequate treatment measure is implantation of a defibrillator-cardioverter for the purpose of primary or secondary prevention of life-threatening arrhythmias and, ultimately, improving the prognosis [2, 74].
Thus, the strategy of therapeutic measures for HCM is quite complex and involves an individual analysis of the entire complex of clinical, anamnestic, hemodynamic parameters, the results of gene diagnostics and risk stratification of VS, an assessment of the characteristics of the course of the disease and the effectiveness of the treatment options used.
In general, rational pharmacotherapy in combination with surgical treatment and electrocardiotherapy allows one to obtain a good clinical effect, prevent the occurrence of severe complications and improve the prognosis in a significant proportion of patients with hypertrophic cardiomyopathy.
For questions regarding literature, please contact the editor.
S. A. Gabrusenko , Candidate of Medical Sciences Yu. V. Safrygina V. G. Naumov , Doctor of Medical Sciences, Professor Yu. N. Belenkov , Doctor of Medical Sciences, Professor of the Research Institute of Cardiology named after. A. L. Myasnikova RK NPK Ministry of Health of the Russian Federation, Moscow
4. Treatment of esophageal spasms
Treatment for esophageal spasm includes treatment of other problems that may worsen or worsen esophageal spasm. For example, GERD is usually treated with changes in diet, lifestyle, and medications that reduce the amount of acid in the stomach.
Here are some things that can help treat GERD and esophageal spasm:
- Eating little and often is better than 2-3 times a day in large portions.
- After eating, wait 2-3 hours before going to bed. Late-night snacking is not the healthiest thing.
- Chocolate, mint and alcohol can worsen GERD. These foods relax the valve between the esophagus and stomach.
- Spicy foods, foods high in acid (tomatoes and oranges), and coffee can also aggravate the problem.
- It is worth giving up smoking.
- Sleeping on an elevated pillow can help relieve GERD at night.
- If you are overweight, you need to lose weight. Losing even a few extra pounds can really help.
Direct treatment of spasms
Esophageal surgery can be done with medications that relax the muscles of the esophagus. But these medications do not always help. Interestingly, some antidepressants can help relieve pain even if you are not depressed. But only a doctor should prescribe them.
Combating stress and anxiety, breathing exercises can also be effective in treating esophageal spasm.
, surgery is required to treat esophageal spasm.
. During the operation, the surgeon cuts the muscle along the lower part of the esophagus. But this procedure is really only performed in serious cases when other methods of treating esophageal spasm do not help.
Achalasia cardia
Clinical manifestations of the pathology are dysphagia, regurgitation and chest pain. Dysphagia is characterized by difficulty swallowing food. In some cases, a violation of the act of swallowing develops simultaneously and is stable; Dysphagia is usually preceded by influenza or another viral disease or stress. In some patients, dysphagia is initially episodic (for example, when eating hastily), then it becomes more regular, making it difficult to pass both solid and liquid food.
Dysphagia with achalasia cardia can be selective and occur when only a certain type of food is consumed. Adapting to impaired swallowing, patients can independently find ways to regulate the passage of food masses - hold their breath, swallow air, wash down food with water, etc. Sometimes, with achalasia cardia, paradoxical dysphagia develops, in which the passage of liquid food is more difficult than solid food.
Regurgitation in achalasia cardia develops as a result of the reverse reflux of food masses into the oral cavity during contraction of the esophageal muscles. The severity of regurgitation can be in the form of slight regurgitation or esophageal vomiting, when profuse regurgitation “mouth full” develops. Regurgitation can be periodic (for example, during eating, simultaneously with dysphagia), occur immediately after eating or 2-3 hours after eating. Less commonly, with achalasia cardia, reflux of food can occur during sleep (the so-called nocturnal regurgitation): in this case, food often enters the respiratory tract, which is accompanied by a “night cough.” Slight regurgitation is typical for stages I – II of achalasia cardia, esophageal vomiting – for stages III – IV, when overflow and overstretching of the esophagus occurs.
Pain with achalasia cardia can be bothersome on an empty stomach or during eating and swallowing. Pain sensations are localized behind the sternum, often radiating to the jaw, neck, and between the shoulder blades. If in stages I–II of achalasia cardia the pain is caused by muscle spasm, then in stages III–IV it is caused by developing esophagitis. For achalasia cardia, periodic paroxysmal pain is typical - esophagodynic crises, which can develop against the background of excitement, physical activity, at night and last from several minutes to one hour. A painful attack sometimes goes away on its own after vomiting or passing food masses into the stomach; in other cases it is relieved with antispasmodics.